
Abstract
Malignant brain tumors such as glioblastoma (GBM) and brain metastasis have poor prognosis despite conventional therapies. Successful use of vaccines and checkpoint inhibitors in systemic malignancy has increased the hope that immune therapies could improve survival in patients with brain tumors. Manipulating the immune system to fight malignancy has a long history of both modest breakthroughs and pitfalls that should be considered when applying the current immunotherapy approaches to patients with brain tumors. Therapeutic vaccine trials for GBM date back to the mid 1900s and have taken many forms; from irradiated tumor lysate to cell transfer therapies and peptide vaccines. These therapies were generally well tolerated without significant autoimmune toxicity, however also did not demonstrate significant clinical benefit. In contrast, the newer checkpoint inhibitors have demonstrated durable benefit in some metastatic malignancies, accompanied by significant autoimmune toxicity. While this toxicity was not unexpected, it exceeded what was predicted from pre-clinical studies and in many ways was similar to the prior trials of immunostimulants. This review will discuss the history of these studies and demonstrate that the future use of immune therapy for brain tumors will likely need a personalized approach that balances autoimmune toxicity with the opportunity for significant survival benefit.
Keywords
Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumor and despite surgical resection, radiation and chemotherapy, overall survival is 14–15 months from the time of diagnosis.1,2 While systemic treatments have improved long-term survival for other solid tumors like melanoma, breast, renal cell and lung, the predilection for brain metastasis in these cancers remains a major limitation for life expectancy. The significant clinical need for effective treatments of brain tumors has pushed the exploration of immunotherapy to treat malignancies into the brain, an area with unique limitations on immune function. While immunotherapy for the treatment of malignancy has expanded in clinical practice over the past two decades, manipulating the immune system to treat cancer, including brain cancer, has a long history that should be considered while developing future therapeutics. In this review, we will highlight the history of treatments designed to augment the effectiveness and efficiency of the immune response against malignant brain tumors, as well as the limitations of autoimmune toxicity. In examining the treatment of primary brain tumors and brain metastasis, we will focus on vaccinations strategies, checkpoint inhibitors and the use of immunostimulants.
Introduction to immunology principles
The role of the immune system is to recognize deviations from normal homeostasis as danger. Most often, this is thought of as protection from infection, however, protection from malignancy may be an even greater focus of human immunity. The development and continued progression of cancer without treatment demonstrates the failures of cancer immune surveillance. Growing understanding of the complexity and highly integrated nature of the immune system is beyond the scope of this review, however, review of basic immune principles and their application to brain tumors is necessary.
Innate immune system
The innate immune system encompasses both the anatomical barriers of our epithelial and mucosal surfaces as well as a surveillance system of cellular components. Natural killer cells (NKs), macrophages, neutrophils, dendritic cells (DCs) and monocytes are capable of phagocytosis, as well as releasing cytokines and chemokines. 3 The innate immune system relies on pattern recognition for specificity. In the case of microbes, receptors are specific for ligands that are uniquely expressed by microbes like lipopolysaccharide or single-stranded DNA, referred to as pathogen-associated molecular patterns (PAMPs). Macrophages and DCs also have receptors for danger-associated molecular patterns (DAMPs) that include heat shock proteins (HSPs) and other signals available during tissue damage and cell death. 4 DAMPs are also released during the development and growth of malignancies. Manipulating PAMPs and DAMPs is one of the broad strategies used in targeting cancer with immunotherapy (Table 1).
Immunostimulants used in cancer theraputics.

BCNU, carmustine; GM-CSF, granulocyte-macrophage colony-stimulating factor; Th1, type 1 T helper cells; IL, interleukin; IFN, interferon; IV, intravenous; IT, intrathecal; LM, leptomeningeal metastasis; rGBM, recurrent glioblastoma; SQ, subcutaneous; TMZ, temozolomide; TNF, tumor necrosis factor; TGF- β, transforming growth factor beta; PAMPs, pathogen-associated molecular patterns; MHC, major histocompatibility complex.
Adaptive immune system
There are bridges between the innate and adaptive immune system, such as the DC and NK cells, which can serve as antigen-presenting cells (APCs) to T cells. 5 Adaptive immunity is more specific and diverse than the innate immune system. However, it requires days of selection and proliferation after initial exposure to an antigen to mount B- and T-cell-based immune response. After initial delayed response, memory T- and B-cell lymphocytes can then quickly respond to previously encountered antigens.
T cells become activated when two criteria are met: (1) the T cell receptor binds an antigen presented by major histocompatibility complex (MHC), (2) the T cell is costimulated by CD28 receptor, binding the ligands of CD80 and CD86 (also known as B7.1 or B7.2, respectively). If a T cell binds an antigen presented by MHC, but also binds a co-inhibitory ligand, or negative checkpoint, this impairs the T-cell response. During normal T-cell activation, there is a balance of costimulatory and checkpoint activation generating a negative feedback loop to dampen immune activation. Regulatory T cells (Tregs) have a central role in maintaining this balance with immunologic tolerance to normal tissues. One-way Tregs downregulate inflammation through expression of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4, also known as CD152), which binds CD28 with higher affinity than CD80 or CD86 blocking the costimulation necessary to activate T cells. 6 Additionally, various cytokines direct T-cell differentiation. For example, interleukin (IL)-2 polarizes to Th1 phenotype, which is more cytotoxic, while IL-4 promotes Th2 phenotype to amplify B-cell production. Alternatively, Tregs secrete Th3 cytokines like IL-10 and transforming growth factor (TGF)-β, which are immunosuppressive. Over the past 20 years, Tregs have emerged as essential components to preventing autoimmunity, but may also lead to malignancy immune tolerance.
Immunology principles related to malignant brain tumors
The brain is an immunologically specialized area, but it is not as immune privileged as once thought. The concept of immune privilege largely originated from an early study in which rabbits did not reject foreign skin grafts to the brain, while skin grafts to other areas were rapidly rejected. 7 It was later shown that these grafts were rejected when grafted to brain and the rejection just took longer than other areas; 8 demonstrating that the brain has active immune surveillance, just in a different form than in other tissues. This immune surveillance can also inappropriately target normal brain as it does in multiple sclerosis, the most prevalent neuro-inflammatory disease of the central nervous system (CNS). 9 Additionally, in systemic tumors the immune system does, rarely, develop an immune response to normal brain antigens in what are known as paraneoplastic syndromes. 10
Many of the immune privilege qualities of the CNS have been attributed to the blood–brain barrier (BBB) which is not an absolute border of protection, but rather limits transit of molecules and helps regulate lymphocyte tracking under normal circumstances. While the brain lacks typical lymphoid tissue, there are lymphatic vessels that drain antigens from the dural sinuses to the cervical lymph node chains. 11 Additionally, antigen presentation functions differently in the CNS with epithelial cells, astrocytes, microglia, macrophages and DCs all acting as potential APCs. The nuances of immunity in the CNS continue to drive stimulating research which will likely introduce additional immunotherapeutic targets. 12
Although the CNS is not as immunologically privileged as once thought, the brain can serve as a sanctuary for tumors that have otherwise responded to systemic immunotherapy chemotherapy. 13 There are several strategies that lead to immune escape in the CNS. GBM provides the best studied example of tumor-associated immune suppression and evasion in the brain. GBM creates immunosuppression both within its microenvironment and also systemically (Table 2). Usually phagocytic, microglia or macrophages compose one third or more of GBM volume. 14 Prolonged exposure to glioma cells alters the normal antitumor response of these immune cells to facilitate immunosuppression.15–17 Within GBM, microglia, as well as glioma cells themselves, secrete potent immunosuppressive factors that inhibit T-cell growth, downregulate major histocompatibility complex class II (MHC II) on APCs and induce Tregs.18,19 In addition to altering the microenvironment, GBM also induces systemic immunosuppression appreciated as lymphopenia and decreased ability to mount T-cell responses with preserved B-cell activity.20–22 Peripheral blood lymphocytes also have deficits in IL-2 signaling and many other glioma-derived immunosuppressive factors are still being investigated. 23
Tumor-mediated Immunosuppression.
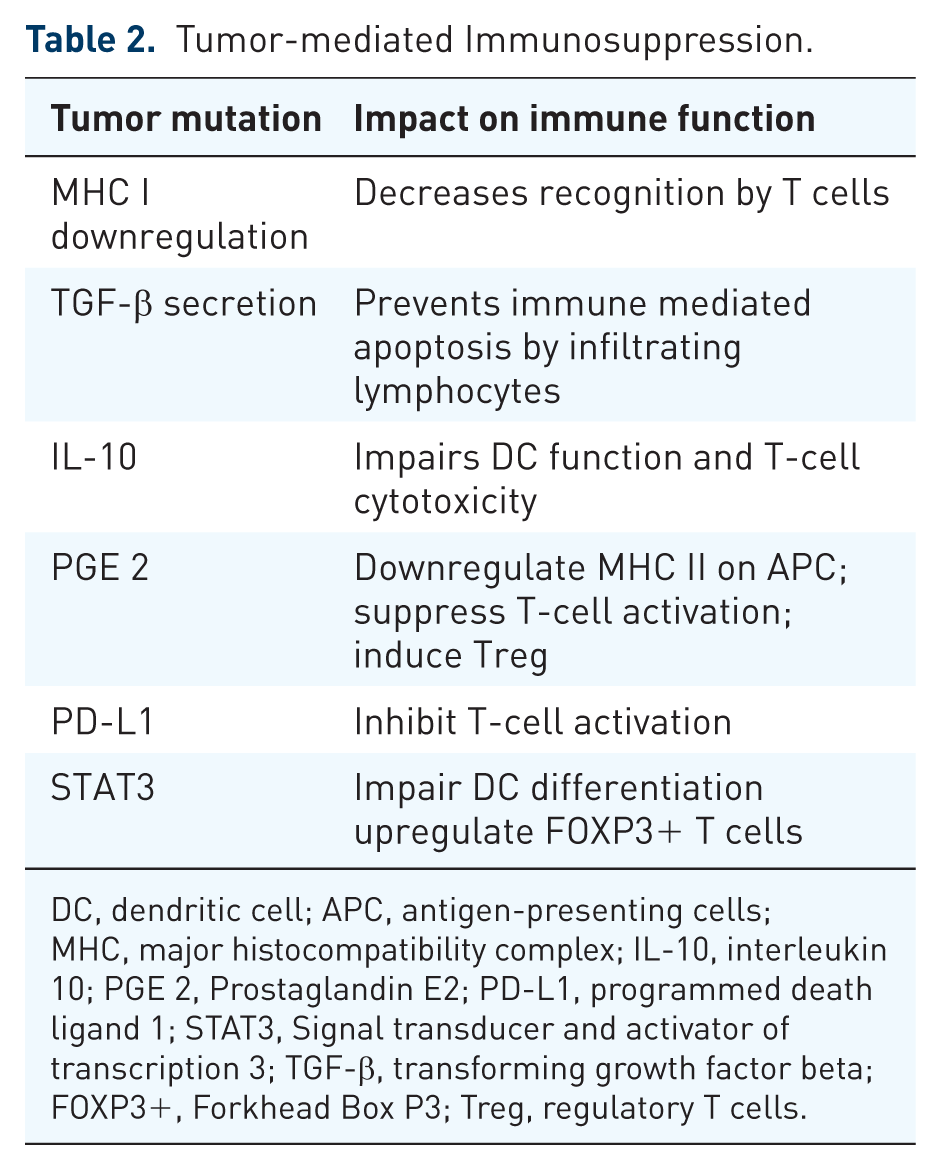
DC, dendritic cell; APC, antigen-presenting cells; MHC, major histocompatibility complex; IL-10, interleukin 10; PGE 2, Prostaglandin E2; PD-L1, programmed death ligand 1; STAT3, Signal transducer and activator of transcription 3; TGF-β, transforming growth factor beta; FOXP3+, Forkhead Box P3; Treg, regulatory T cells.
With expanding understanding of immune system activation and regulation, as well as the CNS specific nuances to immune therapy, there continue to be expanding therapeutic targets for immunotherapy of brain cancer (Figure 1). It is important to consider the history of therapies that have been implemented, as our understanding of the immune system continues to develop.
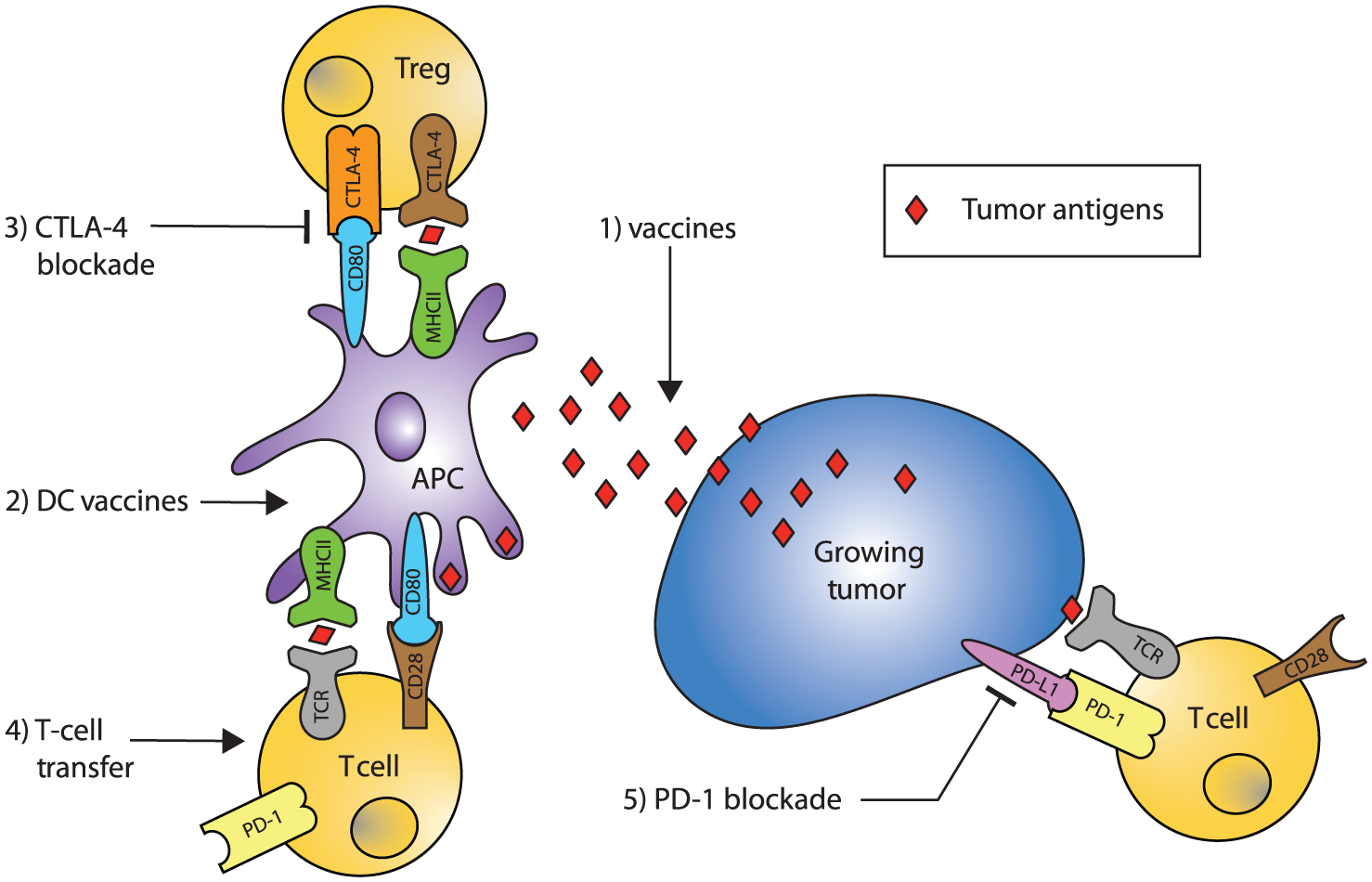
Schematic of immune response to growing tumor and targets of immunotherapy.
Historical perspective
The history of immunotherapeutics for malignancy dates back to the early 1900s, when using antibodies to target cancer was proposed to provide specificity and minimize toxicity. 24 This hypothesis was followed by a number of case reports of spontaneous remission, or remission at distant sites from radiation, providing evidence that the immune system was capable of combating malignancies.25–27 In the 1950s, autoantibodies to tumors were identified and there was growing evidence of immune cell infiltration into various malignancies.28–30 During the 1960s, after decades of disappointment, preclinical studies demonstrated that rodents immunized with irradiated cancer cells were resistant to subsequent challenges with tumor31,32 (reviewed by Srivastava and Old 33 ).
Vaccines
A vaccine is a therapy targeted at acquiring long-term immunity, or an adaptive immune response against antigen(s) of interest. Classically, the benefit of vaccination is in a prophylactic capacity; however, when applied to malignancy, the target is to help initiate antitumor immunity. Attempts at cancer vaccination have taken many forms, from passive immunization with antitumor antibodies to actively generating an immune response with autologous/allogeneic tumor lysate, synthetic peptides, naked DNA or recombinant viral vectors, as well as administering immune cells directly to patients. These various techniques are attempting to identify tumor-rejection antigen(s) and stimulate an effective immune response while avoiding autoimmune pathology and preventing immune evasion. Both peptide and cell-based vaccines have used various strategies: (1) epitopes in conjunction with carrier proteins to enhance immunogenicity, (2) adoptive T-cell transfer and (3) DCs pulsed with peptides.
Tumor lysate
In response to the sentiment that the prognosis for high-grade glioma was ‘hopeless’, investigators rapidly translated a rodent study in a fibrosarcoma model to GBM in humans. 34 Bloom and his team of investigators examined subcutaneous injections of irradiated, non-necrotic tumor to the standard of care of radical surgery and postoperative radiation. There was no survival benefit; no vaccinated patients survived past 30 months when the control group had 7 of 35 patients surviving beyond this point. A companion study in anaplastic gliomas, studied monthly vaccination with glioma cell lines augmented with the adjuvant Bacillus Calmette–Guérin (BCG), but also failed to show a survival benefit. 35 Interestingly, this study had a 20% rate of dementia that was postulated could be autoimmune in etiology, although further investigation was limited.
Given limited success with tumor lysate alone, investigators worked to increase the T-cell response to antigens. One strategy, T-cell transfer, involves first vaccinating patients with irradiated autologous tumor cell, followed by inguinal lymph-node biopsy to harvest the T cells that respond to the vaccine. These cells were then expanded in vitro and transfused back into the patient as an adoptive transfer of activated T cells. GBM systemic immunosuppression did limit the T-cell harvest, however this small study had promising survival for recurrent glioma of 12 months compared with historical controls of 6 months. 36 This outcome was despite 5 of 10 patients with marked increase in tumor size after treatment. Adoptive T-cell transfer has continued to be refined and demonstrated tumor regression in melanoma brain metastasis. 37 Autologous T-cell vaccination is limited by T-cell dysfunction in GBM patients and is also inherently complex and expensive, which led to using an alternative vaccination strategy targeted at stimulating T cells.
DCs can furnish all the signals needed to stimulate native T cells and initiate an adaptive immune response. A professional APC, DCs can be isolated directly from blood by negative selection or cultured from progenitor cells. Beginning in the 1990s, there was a surge of great work published using this technique in rodent intracranial tumor models. These studies cultured resected tumor cells in vitro and used various preparations to optimally stimulate DCs. The outcomes were promising with prolonged survival and evidence of T-cell-mediated antitumor response in rodent cancer models.38–41 Dr. Liau’s group expanded this work to a dose-escalation clinical trial of DCs stimulated with autologous tumor-associated proteins in patients with GBM. 42 Surprisingly, there was no correlation between systemic antitumor response and clinical response, even though 50% of patients (four of eight) who underwent reoperation for recurrence demonstrated T-cell infiltration into the tumor after vaccination. Similar safety, systemic antitumor response and T-cell infiltrate into tumor recurrence were reported by another group. 43 This work has led to multiple clinical trials, including the soon-to-be-completed phase III DCVax-L (also known as DCVax-Brain) which included expanded access for patients who do not meet the phase III enrollment criteria [ClinicalTrials.gov identifier: NCT02146066]. This technology has the advantage of targeting multiple antigens and facilitating T-cell activation in a cancer with impaired T-cell function, however, success of this strategy will likely require further patient selection to result in clinical benefit.
Heat shock protein-96
HSPs are an abundantly expressed group of molecules with highly variable expression that is increased under heat shock as well as other stresses that include the high metabolic demand of tumor cells. These intercellular chaperones were identified in the 1980s to be DAMPs and have a unique role in generating specific immune responses. HSPs function as a selective tumor-lysate antigen-delivery molecule. Normal tissue HSPs cannot elicit immunity, however, the complex of HSP and associated tumor antigens can elicit protective immunity. 44 The endoplasmic reticular HSP, HSP-96, was found to be particularly effective at activation and maturation of DCs and many of the identified peptide vaccine targets mentioned below are known substrates of HSP-96. 45 Intradermal injection of autologous tumor HSP-96 vaccine was used in a phase I and a multicenter phase II trial in recurrent GBM with 90.2% of patients surviving longer than 6 months.46,47 While this technique had limited success in renal cell carcinoma and melanoma, there remains hope for benefit in selected patient populations with GBM.48,49 There is an ongoing randomized phase II trial of HSP-96 vaccine in combination with bevacizumab either initially, or at progression, compared with bevacizumab alone in recurrent GBM [ClinicalTrial.gov identifier: NCT01814813]. While not yet reported, this vaccine was also studied in 46 patients with newly diagnosed GBM [ClinicalTrial.gov identifier: NCT00905060]. Other autologous HSP-96 vaccines are currently in phase I studies for new diagnosis GBM [ClinicalTrial.gov identifier: NCT02122822] and pediatric glioma [ClinicalTrial.gov identifier: NCT02722512]. HSP-96 vaccines provide the benefit of multiple antigens of tumor lysate, but with a more refined selection of immunogenic antigens capable of activating the innate immune system.
Peptide vaccines
Recognizing that tumors have profound genomic, transcriptomic and proteomic alterations, many studies have worked to identify tumor-associated and tumor-specific antigens. While nearly 10,000 potential targets have been identified, fewer have the ability to activate the immune system or serve as immunogenic antigens. Antigens that meet both of these criteria are divided functionally into tumor-specific antigens and tumor-associated antigens. Tumor-specific antigens are not expressed in normal tissues and are often the product of mutations or splice variants (examples for GBM include EGFRVIII and IDH-1). More common are tumor-associated antigens, which can be viral antigens, over expressed or amplified gene products (EGFR, survivin, EphA2, IL-13Rα2), differentiation antigens (gp 100, WT1) or antigens usually restricted to germ cells that are expressed in tumor cells (MAGE-1, MAGE-3). EGFRvIII is the prototypical tumor-specific antigen studied in GBM while viral and other tumor-associated antigens are often studied in therapies targeting multiple antigens.
Epidermal growth factor receptor vIII
Epidermal growth factor receptor variant type III (EGFRvIII) is a mutation found in 15–60% of primary GBM and lower percentages in breast, lung, prostate and colorectal cancer.50–52 This mutation is not found in normal tissues and serves as the best-studied example of a tumor-specific antigen in GBM. This mutation creates a ligand independent, constitutively active tyrosine kinase that has been shown to augment proliferation, inhibit apoptosis, promote tumor-cell motility and confer resistance to radiation, which has suggested a direct oncogenic effect.53–56 In the selection of GBM patients who had gross total resection and survived past 12 months, EGFRvIII expression has been suggested as an independent negative prognostic factor.57,58
A 14 amino-acid portion of the EGFRvIII peptide containing the novel glycine and terminal cysteine was conjugated to the carrier protein keyhole limpet hemocyanin (KLH), creating PEPvIII-KLH. This peptide has been used as the peptide for several clinical trials after DCs pulsed with it demonstrated efficacy against U87 glioma cells. 59 The VICTORI trial used EGFRvIII-peptide-pulsed DCs in a dose-escalation trial in 12 patients with new diagnosis GBM. 60 Authors expressed an appropriate concern about the possibility of devastating toxicity if there was cross-reactivity with normal brain antigens, however, they reached the technically limiting concentration of DCs without significant toxicity. Notably, patients were not screened for EGFRvIII expression in this trial. The study also examined systemic immune responses and while in breast cancer studies, some patients demonstrated immune response to EGFRvIII prior to vaccination, none of the GBM patients did in this, or subsequent GBM trials. 61
Investigators then pivoted away from the use of DCs, favoring intradermal peptide injections with Granulocyte-macrophage colony-stimulating factor (GM-CSF) as adjuvant. Peptide preparations have the benefit of a more standardized therapy, reduced cost and ease of production when compared with cell preparation techniques. The ACTIVATE trial studied patients with new diagnosis GBM who were screened for EGFRvIII expression. The 18 vaccinated patients had an overall survival of 26 months, which compared favorably with matched historic controls. 62 Perhaps the most significant finding from this study was that 9 of the 11 recurrent tumors that were pathologically evaluated had lost EGFRvIII expression at recurrence. This supports that vaccination resulted in immune system recognition and clearance of this antigen. Notably, this immune response did not result in significant treatment-related toxicity or excessive inflammation. In light of temozolomide (TMZ) becoming the new standard of care in 2005, 2 there was concern that the lymphopenic state induced by TMZ would impair vaccine efficacy. To the contrary, there was building preclinical data that lymphopenic states could induce autoimmunity or augment antitumor immune response driving away from Treg and more towards a cytotoxic T cell response.63,64 ACTII sought to evaluate EGFRvIII vaccination in combination with TMZ therapy. Consistent with preclinical data, there was a more enhanced cellular and humoral immune response in the TMZ dose-intensified group, which had more sustained lymphopenia. This, however, did not alter progression-free survival or overall survival compared with the standard dose group (although the study was not powered to detect a difference). This study did begin to test the balance of immune activation with autoimmune-based toxicity, as there were allergic drug reactions in 4 out of 10 patients, including one who was removed from the study with cardiovascular adverse events. 65 Again, 11 out of 12 pathologically evaluated recurrent tumors had lost expression of EGFRvIII. A finding that was confirmed in the limited number of recurrent tumors obtained from the multicenter phase II, ACT III that also validated prior overall survival and progression-free survival benefit compared with historic controls. 66 Given continued promising results of these open label studies, ACT IV, a double-blind phase III trial, began enrollment. In a press release in early 2016, Celledex, the company sponsoring the study, announced they were discontinuing this study, based on interim analysis where the control arm significantly outperformed expectations with a 21.1 month overall survival compared with 20.4 months in the vaccine group. 67 Despite this surprising result, there continues to be interest in EGFRvIII. ReACT, a randomized phase II study of EGFRvIII peptide vaccine in combination with bevacizumab in patients with recurrent GBM, is the only study to date to show a statistically significant increase in overall survival from 9.3 months in control to 11.3 months. This was accompanied by a robust immune response to EGFRvIII that did correlate with improved outcomes. There was also a subset of patients with radiographic response greater than 18 months and survival for some patients exceeding 30 months. 68
EGFRvIII is also being examined in chimeric antigen receptors (CARs). CARs are synthetic molecules that consist of a fusion of the extracellular variable domain of monoclonal antibodies fused to intracellular T-cell signaling domain. CARs are genetically expressed in T cells and mediate potent antigen-specific, MHC-independent activation of T cells to recognize tumor. 69 This technology is particularly promising for cancers with MHC downregulation, like GBM. While this technology has shown promise in other solid tumor malignancies like neuroblastoma and renal cell carcinoma, there have also been significant adverse events from cytokine release syndrome, unexpected organ damage and significant neurotoxicity, as well as patient deaths.70–73 In a preclinical GBM mouse model, EGFRvIII CAR-T cells effectively migrated to intracranial tumor and demonstrated improved overall survival compared with untreated and control CAR-T animals. 74 EGFRvIII CAR-T cells were humanized and specificity was verified in human xenograft models prior to development of clinical trials for GBM [ClinicalTrial.gov identifiers: NCT02209376 and NCT02664363]. 75 Additional work has also been directed at adjusting the duration of expression to decrease of tissue toxicity. 76 There are several other antigens that have been targeted using CAR-T in mouse models of GBM, including IL-13Rα2, HER2 and EphA2.77–79 While CAR-T cells are limited in the scope of antigens they can target, the ability to recognize tumors in an MHC-independent manner may prove to be a more effective therapy for immunosuppressive tumors.
Of thousands of tumor antigens identified in GBM, EGFRvIII remains the prototypical tumor-associated antigen for GBM vaccine studies. The work to date has not only validated the mechanism of vaccination with the eradication of EGFRvIII expression during recurrence, but also demonstrated the limitations of targeting a single antigen in a heterogeneous disease. EGFRvIII work demonstrated the careful balance of safety when activating the immune system in brain cancers, with rare autoimmune toxicity, perhaps a benefit of targeting a tumor-specific antigen.
Viral antigens
Cytomegalovirus (CMV) is a beta-herpes virus that infects 50–90% of the adult population but only causes encephalitis in fetuses and immune-compromised patients. It has been detected in several human malignancies and it remains unclear if this is local reactivation or if CMV plays a role in pathogenesis of GBM.80–82 Preclinical evidence demonstrated CMV proteins deregulate multiple cellular pathways increasing cellular proliferation, angiogenesis and immune evasion. 83 While a role in oncogenesis is unclear, a randomized control trial of valganciclovir in addition to standard of care therapy for 6 months did not increase survival in GBM, although a retrospective analysis of longer treatments suggested a prolongation.84,85 While a primary pathogenic role is unclear, the high percentage of CMV gene products expressed in malignant gilomas, but not in surrounding brain tissue, make CMV antigens good targets for antitumor vaccines.
Phosphoprotein 65 (pp65) is a dominant CMV epitope with reported expression in new diagnosis GBM varying from 50% to 70%. Interestingly, in one of the above-described autologous tumor-lysate DC vaccine studies, a patient developed a robust T-cell response to pp65. 86 CMV antigens have also been targeted using CMV-reactive autologous T cells that were expanded in vitro and infused into patients with recurrent GBM in a phase I/II trial with a median overall survival of 13.4 months. 87 One patient who was pathologically evaluated at disease progression, demonstrated tumor-infiltrating CMV-specific T cells that had higher expression of checkpoint inhibitory receptors, programmed death (PD)-1 and CTLA-4 than T cells from the peripheral blood, supporting a tumor microenvironment shift to Tregs. Although a single case, this also suggests that while these antigens are sufficient to mount a T-cell response, the activity of those T cells in the tumor microenvironment may not be antitumor. A small placebo-controlled study of newly diagnosed GBM patients used preconditioning with tetanus/diphtheria toxoid followed by DCs pulsed with pp65 in six patients compared with tetanus/diphtheria toxoid alone. 88 This small study demonstrated marked increase in survival with 50% survival at 40 months and zero in the toxoid only group. This has generated enthusiasm for ongoing trials to target pp65, as well as other CMV-associated antigens including immediate early 1 (IE1) and glycoprotein B (gB).
Tumor-associated antigens
There are many overlapping tumor-associated antigens across multiple malignancies, however, there remains significant heterogeneity in expression, not only within same category of malignancy, but also within individual tumors. Recognizing this heterogeneity and the proven limitations of a single antigen target, there have been various strategies for multiple antigen vaccines, ranging from off the shelf, to customized or utilizing peptide vaccines to augment autologous tumor-lysate strategies. For the strategies that have translated to clinical studies, there are variable successes. The majority of this work has been targeting malignant gliomas, as vaccination strategies for other malignancies have excluded patients with CNS involvement. There was one patient in a small trial of autologous T cells stimulated by MART1 in malignant melanoma that had brachial plexus metastasis that improved with vaccine treatment. 89
One of the first clinical attempts to vaccinate GBM patients used in vitro prescreening of their immune response to 20 antigens to develop customized vaccines for each patient. In the patients with partial response or stable disease after vaccination, there was in vivo evidence of immune response (measured as T-cell response, IgG production or delayed-hypersensitivity skin testing). 90 However, some patients with progressive disease had similar immune responses after vaccination. Differing from the EGFRvIII work that histologically demonstrated loss of EGFRvIII staining in recurrence, in the limited samples from a study of low-grade glioma immunized to IL13Rα2, EphA2, WT1, and survivin, recurrence demonstrated robust immunostaining for these antigens. 91 This suggested that these vaccinations may be shifting the GBM microenvironment towards Treg phenotype, despite the use of adjuvants such as polyinosinic–polycytidylic acid stabilized by lysine and carboxymethylcellulose (poly-ICLC) to augment a cytotoxic T cell response.91,92
The first immunotherapy to demonstrate a statistically significant improvement in progression-free survival in a phase II study of newly diagnosed GBM was ICT-107, a multiple-antigen-pulsed DC vaccine containing multiple tumor-associated antigens (HER2, TRP-2, gp100, MAGE-1, IL13Rα2, and AIM-2). In the initial phase I trial, there was a trend for vaccine response that did correlate with better survival in patients whose tumors were known to express at least three of the antigens. 93 The phase II study in newly diagnosed GBM did not show an overall survival benefit, but did show 2–3 months progression-free survival that was statistically significant compared with patients treated with DCs that were not pulsed with antigens. 94 There is an ongoing phase III trial [ClinicalTrial.gov identifier: NCT02546102] studying this vaccine in newly diagnosed GBM with the primary outcome of overall survival. Screening for expression of these antigens was not a requirement for enrollment in the phase II or III trials, however, in preliminary analysis, expression of four targeted antigens was associated with prolonged survival. 95
An alternative strategy, Gliovac, has created a protocol addressing many of the limitations from prior studies. Gliovac uses a combination of autologous tumor lysate and allogeneic antigens, as well as the adjuvant GM-CSF, but also pretreats with low-dose cyclophosphamide to deplete Tregs to favor the development of cytotoxic T cells. 96 This small study of nine patients with recurrent GBM had a 100% survival at 6 months, where historical control survival is 33%. This vaccine is currently undergoing phase II trial in new diagnosis GBM [ClinicalTrial.gov identifier: NCT01903330].
This collection of studies supports the need for multiple antigens to optimally target aggressive, immunosuppressive brain tumors like GBM. While there is logistical benefit from the use of peptide vaccines, it remains unclear if this is equivalent to the more challenging and variable cell-based approaches of autologous T-cell transfer and DC vaccines. Increasing clinical experience with DC vaccines now that sipuleucel-T, a peptide-stimulated DC vaccine for prostate cancer, is FDA approved, may further support the clinical use of cell-based therapies over peptide vaccines. Although trials with EGFRvIII support the ability of peptide vaccination to target and eliminate selected cells, or at least antigens from GBM, recurrence indicates the need for multiple antigens. Additionally, continued work to identify biomarkers to guide patient selection for successful use of vaccine strategies will be essential. Overall, vaccine therapies have been well tolerated with most common response of transient flu-like symptoms. This is in contrast to the use of immune-checkpoint inhibitors that were initially developed to augment vaccination strategies, but demonstrate a broad and relatively toxic side-effect profile for patients.
Immune checkpoints
The emerging manipulation of immune-cell checkpoints to treat malignancy has rapidly advanced the treatment of multiple malignancies, however, further pushed the boundary of autoimmune toxicity. These agents act by blocking the immunosuppressive receptors that inhibit cytotoxic T cells and increase the antitumor response. Many of the initial studies of these therapies excluded patients with CNS pathology, however, there is increasing evidence brain tumors, both primary and metastatic, respond to these treatments. These agents have also introduced a new challenge with considerable toxicity related to the immune-system response to non-neoplastic cells in an unpredictable manner. CTLA-4 and PD1/PD-L1 are two negative regulatory pathways of T cells that have FDA-approved therapies, with several others in the preclinical and early clinical pipeline.
Cytotoxic T-lymphocyte-associated antigen 4
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4, also known as CD152) is an inhibitory molecule that blocks T-cell activation by binding CD80 and CD86 (also known as B7.1 and B7.2) with higher affinity than the T cell costimulatory receptor CD28. It is highly expressed by Treg and is vital to their inhibitory function. The importance of CTLA-4 in balancing T-cell activation and inhibition is demonstrated by the CTLA-4 knockout mouse that develops fatal systemic autoimmunity from unopposed T-cell activation to self-antigens. Blocking CTLA-4 drives cytotoxic T-cell activity, resulting in breaking this natural protection from autoimmunity.
In 2011, the FDA approved a fully human IgG antibody directed against CTLA-4, ipilimumab, for the treatment of unresectable or metastatic melanoma. Pooled analysis of long-term survival data in metastatic melanoma support a durable effect in some patients that has been sustained up to 10 years, a remarkable outcome for a diagnosis with a historical survival of 8–10 months. 97 Prior to the FDA approval of ipilimumab, reports were suggesting benefit for this therapy in CNS metastasis of melanoma. A dose-escalation study of ipilimumab was undertaken in combination with peptide vaccines to gp100. The expectation of this study was that checkpoint blockade would augment the immune response as it had in preclinical studies; however, this study demonstrated that ipilimumab alone was sufficient to elicit an antitumor effect that was greater than the peptide vaccine alone or the combination therapy. 98 Encouragingly, this early study included one patient with brain metastasis who demonstrated a complete CNS response at 31 months. 99 This study was also the first to report a correlation between immune-related adverse events and clinical response, supporting the idea that the mechanism of action is blocking of immune tolerance to self, as well as cancer. Subsequently, the case of a 63-year-old woman with refractory progressive CNS melanoma involving the spinal cord and brain, who was treated with ipilimumab on a compassionate-use basis was reported. 100 She had treatment complications of fluctuating edema, ultimately requiring treatment with dexamethasone, however, had continued improvement in her performance status until 7 months after treatment. With the recurrence of focal seizures, she underwent surgical resection that demonstrated activated T-cell infiltrate into the left frontal mass. This detailed case describes the most common CNS-related adverse events of cerebral edema and seizures and supported CNS immune response with this therapy.101,102
While immune-related adverse events developed in the majority of patients in these early trials (62% or 86 of 139 patients), there were minimal reports of CNS-related immune toxicity, even though the trial included 10 patients with CNS metastasis. 103 In the first phase III trial, 10–15% of patients experienced grade 3 or 4 immune-related adverse events (most commonly skin and gastrointestinal) and there were seven deaths associated with immune-related adverse events. 98 Headache was the only CNS symptoms. When ipilimumab was used in combination therapy with dacarbazine, the immune toxicity significantly increased to 56.3% of patients experiencing grade 3 or 4 adverse events (of note, this trial excluded patients with evidence of brain metastasis). 104
Given toxicity and concern for significant complications, if an uncontrolled immune reaction was initiated in the brain, an open-label trial specifically studied the safety and activity of ipilimumab in a population with active CNS metastasis. They divided the study into two groups: (1) patients taking corticosteroids (n = 21) and (2) patients asymptomatic from brain metastasis and not taking corticosteroids (n = 51). 105 Similar to other immunotherapy studies, enrollment was limited to patients naïve to immunomodulatory therapy and without active autoimmune disease. There were less grade 3 and 4 adverse events in the group on steroid treatment, while overall adverse events were comparable with earlier studies. It remains unclear if steroids impair the efficacy of ipilimumab, as reports of long-term benefit have been in asymptomatic brain metastasis. 106
Additional work has aimed at identifying optimal combination therapy of radiation and ipilimumab, as well as recognition of adverse events in active brain metastasis. A retrospective analysis of 77 patients from one center, reported ipilimumab in combination with stereotactic radiosurgery (SRS) was associated with increased median survival from 4.9 to 21.3 months and an increase in 2-year survival from 19.7% to 47.2%, even when adjusting for performance status. 107 This retrospective analysis also described significant complications with SRS following ipilimumab administration that included necrosis and symptomatic cerebral edema. Conversely, a retrospective study of 58 consecutive patients from a different center examined SRS with or without ipilimumab treatment and found no difference in local control or development of new brain metastasis with ipilimumab compared with SRS alone. 108 Interestingly, this study also reported no radiation necrosis or steroid dependence in patients. Retrospective analysis is working to identify the selected populations that would benefit from ipilimumab.109,110 Additionally, determining the optimal timing for radiation therapy is necessary, as a recent work suggests that timing of SRS within 4 weeks of administration of ipilimumab results in greater radiographic lesion response. 111
Expansion of the use of this therapy to primary brain tumors, specifically GBM, may require combination with other therapies. CTLA-4 blockade has been used in mice to augment vaccination strategies for glioma, similar to its initial use in melanoma studies. Early CTLA-4 blockade alone was associated with increased survival; however, the response was more durable when CTLA-4 blockade was used to augment tumor-lysate vaccines in a mouse model of GBM. 112 A preliminary single group study of ipilimumab in combination with bevacizumab in a mixture of GBM patients has been reported with no grade 4 adverse events but 2 out of 20 patients stopping treatment due to adverse events. 113 While a small study, only 6 of 20 had disease progression at 3 months which is encouraging for the use of this therapy for GBM and current studies are ongoing [ClinicalTrials.gov identifier: NCT02311920].
Programmed death axis, programmed death-1/programmed death-ligand 1
An additional promising target for immune-checkpoint blockade is targeting the association of PD-1 with one of its ligands, PD-L1. PD-1 is a cell surface trans-membrane co-inhibitory receptor on cytotoxic T cells and plays a crucial role in immune escape mechanisms. It reduces T-cell activity, inducing tolerance and decreasing autoimmunity. PD-1 ligands are PD-L2, which is present in antigen-presenting cells, and PD-L1, which is induced in normal cells by inflammatory signals of IFN-γ and TNF-α. PD-L1 expression is generally low in the brain, however, highly expressed in GBM. 114 Recent work identified variable expression in brain metastasis with highest expression in renal cell carcinoma and melanoma, however, expression in these brain tumors was far less than in GBM. They also determined higher expression in smaller brain metastasis than larger tumors. 115 There are currently two PD-1 inhibitors, pembrolizumab and nivolumab, approved for the treatment of melanoma and non-small cell lung cancer (NSCLC). There is one PD-L1 inhibitor, atezolizumab, approved for NSCLC and urothelial carcinoma. Pembrolizumab has been compared directly with ipilimumab in a phase III trial and confirmed superior response and survival in melanoma with PD-1 blockade to CTLA-4 blockade. 116 In this study, there was a strong association between PD-L1 expression in tumors and clinical response, suggesting that PD-L1 may be used as a biomarker for patient selection. With success in systemic disease, there is growing evidence supporting the use of PD-1 inhibitors in brain metastasis and possibly primary brain tumors.
Similar to other agents, early trials using PD-1 inhibitors did not include patients with active brain metastasis, however, there were encouraging case reports of response of brain metastasis from renal cell cancer and melanoma.117,118 There were also warning signs that PD-1/PDL-1 inhibitors should be used with caution. In a case study of 32 patients with NSCLC treated with nivolumab, 12 (37%) had severe events, leading to treatment discontinuation (eight of whom had CNS metastasis). 119 The most common events were severe neurologic symptoms, including altered consciousness, gait disorder, nausea and headache. On average, these symptoms were seen 2–42 days from initiation of nivolumab. This experience was very different from a retrospective study of 26 patients with metastatic melanoma treated with nivolumab, as well as stereotactic SRS where only one patient reported neurologic toxicity, a headache. 120
An open-label phase II trial examined pembrolizumab in asymptomatic, untreated brain metastasis of melanoma and NSCLC. This study reported activity concordant with systemic response, as radiographic response was seen in 4 of 18 patients with melanoma and 6 of 18 with NSCLC. 121 A complete response was reported in four patients with NSCLC, which appears to be a durable response for most, and even a patient with radiographic progression was continued on trial, due to clinical benefit at 20 months. Of note, most patients were treated with prophylactic antiepileptic medications because there was a seizure in one of the early patients. Interestingly, biopsy of presumed tumor progression in one melanoma patient demonstrated inflammation, and not tumor progression, after just the first dose with pembrolizumab, complicating the identification of progression. There are ongoing trials to specifically examine anti-PD-1 therapy in brain metastasis from melanoma and NSCLC [ClinicalTrials.gov identifier: NCT02085070].
Given high expression of PD-L1 in GBM, there has been obvious excitement around the use of PD-1 inhibitors in these patients. In a mouse model of GBM, there was only a survival benefit in animals receiving the combination of PD-1 antibodies with radiation (from 25 to 53 days), but not to PD-1 inhibition alone. 122 Because of broad clinical availability, however, many GBM patients have been treated with these antibodies. Clinical and radiographic response to nivolumab has been reported in two cases of children with recurrent multifocal GBM (associated with mutation biallelic mismatch repair deficiency syndrome). 123 Both children were stable at age 9 years and the other at 5 months, as of publication. At the Society for Neuro-Oncology meeting 2016, early results of 26 patients with recurrent GBM with PD-L1 expression treated with pembrolizumab reported 45% 6-month survival, however, had a subset that maintained durability for up to 80 weeks. 124 Early results of 32 patients with recurrent GBM treated with a PD-L1 inhibitor, durvalumab, found the 6 patients who were progression free at 6 months, remained so for at least 1 year (MEDI14736). The final results of these and other ongoing phase I and II trials studying PD-1 or PDL-1 blockade in relapsed and newly diagnosed GBM, as well as intrinsic pontine glioma are awaiting publication.
Combination therapy
Given the complementary mechanisms of action and promise from clinical practice with sequential therapy of CTLA-4 and PD-1 blockade, active work is underway to examine if combination therapy may be more efficacious. Preclinical studies in mice suggest that PD-1 inhibition is more effective than PD-L1 which is more effective than CTLA-4, however, the combination of CTLA-4 with PD-1 resulted in cure of 75% of animals. 125 Combination therapy was effective with advanced, later-stage tumors and resulted in immune memory preventing tumor rechallenge in rodent models. Unfortunately, when translating to clinical studies, the autoimmune toxicity increased in combination therapy. Checkmate 143 is a phase III study of nivolumab alone or in combination with ipilimumab (Table 3). The most recent preliminary data on this study demonstrated 9 of 10 patients treated with standard-dose ipilimumab in combination with nivolumab experienced grade 3 or 4 adverse events with 4 of 10 patients discontinuing treatment due to side effects.126,127 A nonrandomized group of 20 patients with decreased dose of ipilimumab (1 mg/kg) in combination with nivolumab demonstrated better tolerance with 25% grade 3 or 4 adverse event, however, still increased compared with nivolumab alone. While treatment numbers are small, there were no patients with partial or complete response in either combination treatment group. There is also an ongoing trial for new diagnosis GBM (and gliosarcoma) comparing TMZ in combination with (1) nivolumab or (2) ipilimumab or (3) nivolumab and ipilimumab [ClinicalTrials.gov identifier: NCT02311920]. There is also a phase I study combining nivolumab with a DC vaccine for malignant glioma [ClinicalTrials.gov identifier: NCT02529072].
Ongoing Phase III immunotherapy trials for brain tumors.

DC, dendritic cell; EGFRvIII, epidermal growth factor receptor variant type III; GBM, glioblastoma; IFN, interferon; TMZ, temozolomide.
Immune-related adverse events
A key challenge to the clinical use of these checkpoint inhibitors is the balance of immune response and inflammatory or autoimmune response both locally and off target. There has always been a high level of caution for inflammatory reactions in brain tumors because of the risks of cerebral edema and increased intracranial pressure associated with tumor or inflammation. There are multiple reports of autoimmune neurologic side effects of checkpoint inhibitors including hypophysitis, encephalitis, demyelinating polyneuropathy and encephalomyelitis.128–135 These can often be completely reversible, particularly if recognized and treated early, however several deaths secondary to autoimmune toxicity have been reported. There have also been two case reports, one with melanoma and the other with NSCLC, who 1–2 months after treatment with PD-1 inhibitors had biopsy-confirmed delayed radiation-induced vasculitic leukoencephalopathy. 136 Authors postulate that checkpoint blockade accelerated the immunologically mediated radiation changes in surrounding normal brain tissue. Data so far support that PD-1 blockade is associated with less autoimmune toxicity than CTLA-4 targeted therapy; however, use of PD-1 prior to CTLA-4 blockade or in combination is associated with more toxicity. 137 While the morbidity and mortality from these off-target effects is significant, at least in the metastatic melanoma population, autoimmune toxicity does correlate with clinical response. 99 This supports the idea that the mechanism of action is breaking self-tolerance and stresses the importance of management of these toxicities. Based on the experience from melanoma studies, there are guidelines to suggest median time for appearance of specific toxicities and work up of common autoimmune toxicities. 138 Given the relatively low number of patients with CNS tumors that have been treated, there are not standing recommendations for managing CNS toxicity, however, many recommend obtaining an MRI brain at 1 month after treatment to monitor for progression and edema.
Immunostimulants
Prior to the identification of checkpoint inhibitors, there was a long history of using different agents alone, or in combination with other immunotherapies, to stimulate the immune system to fight malignancy. The use of these agents has also been limited by significant toxicity, including deaths. 139 These agents are generally cytokines, but also include PAMPs used as adjuvants such as BCG or poly-ICLC to augment vaccines. Cytokines are often divided into categories by their association with T-helper phenotypes. The Th1 class stimulates cell-mediated immune response and includes IL-2, IL-12 and interferon (IFN)-ϒ. Th2 class includes IL-4 and IL-10, which have roles in humoral immunity. Th3 class are immunosuppressive cytokines including TGF-β that tumors, like GBM, use to facilitate immune suppression. Cytokines have been administered systemically or locally to direct antitumor activity, as well as to augment other immune therapies. Several cytokines have shown promise in preclinical studies, however there was little benefit and significant neurotoxicity in clinical trials (immunostimulants and sample studies are included in Table 1). We will briefly discuss IL-2, the first and perhaps best studied Th1 cytokine for multiple brain tumors including GBM and melanoma.
IL-2, previously known as T-cell growth factor, is a 15kD glycoprotein that activates cytotoxic T cells, macrophages and B cells, as well as enhances NK cell activity, and stimulates TNFα and IFN-ϒ. IL-2 has been frequently used in vitro to activate immune cells, particularly lymphokine-activated killer cells (LAK), which are autologous lymphocytes stimulated in vitro with IL-2 to generate large granular lymphocytes that have non-MHC restricted cytotoxicity. Alternatively, IL-2, when injected directly into tumors, has been shown to attract lymphocytes into the typically immunosuppressive microenvironment.
Intravenous IL-2 was approved for the treatment of metastatic melanoma and metastatic renal cell carcinoma in the late 1990s. Systemic use is complicated by toxicity related to increased vascular permeability leading to extravasation of fluid resulting in vascular leak syndrome (VLS) and resulting multiorgan impairment. 140 Neurologic symptoms and neuropsychiatric effects have been reported, but it is unclear if this is secondary to VLS or a direct effect of IL-2 on the brain.141,142 Due to these toxicities, this therapy is usually reserved for patients with excellent performance status, and with rare exceptions, excludes patients with brain metastasis. 143
To penetrate the BBB, high doses of IL-2 are required because cerebrospinal fluid (CSF) concentrations are 50% that of serum. 144 The ability to achieve adequate CNS concentrations is limited by the significant systemic toxicity. This lead to the use of intrathecal or intraventricular IL-2 for treatment of CNS disease, primarily for leptomeningeal disease. Based on a single case of a patient who died 50 hours after treatment with intrathecal IL-2, the cellular response was only evident a few millimeters into the parenchyma adjacent to the ventricles. 145 There have been multiple reports of CSF clearance in leptomeningeal disease of melanoma, however, there remained high toxicity and less response in other malignancies including glioma and meduloblastoma.146–149 Acute toxicity, including seizures, headache and fatigue were, in part, related to the rapid increase in intracranial pressure that developed 30–180 minutes after use. 150 There have also been reports of delayed CNS toxicity of dementia, ataxia and white matter changes that are concerning for autoimmune pathology.151,152 Given the significant toxicity, intratumoral or intracavitary use was explored for GBM. In a small study of recurrent GBM, injection of IL-2 alone resulted in an increase in tumor burden, as well as edema. 153 Studies using IL-2 alone, or in combination with autologous cell transfer, reported widely variable toxicity ranging from 100% of patients to no toxicity.154,155 Given CNS toxicity of IL-2, even with direct tumor or tumor-cavity applications, studies used lower doses of IL-2 in combination with autologous lymphocytes that were stimulated in vitro with IL-2 to generate LAK cells. These studies continued to report morbidity and mortality with modest clinical response.154,156,157 The most encouraging report of improved long-term survival was in small group of patients with recurrent GBM or anaplastic astrocytomas with median survival of 12.2 months. 158 This study involved cycles of injections of LAK and IL-2 through Ommaya reservoir followed by five additional injections of IL-2 alone before the next cycle beginning with injection of LAK and IL-2. Overall, the use of LAK with only in vitro IL-2 stimulation has been better tolerated and shown possible clinical benefit when used in larger studies of GBM (33 patients studied with 75% 1-year survival) and recurrent GBM (40 patients with 34% 1-year survival).159,160 Various other strategies to deliver IL-2 have been investigated, primarily in preclinical work, including gene therapy, cytokine secreting cells, biodegradable polymers and retroviral-producing cells.161,162 Results of recently completed intratumoral cell-transfer therapy using low doses of IL-2 to assist with transferred cell viability have not yet been reported [ClinicalTrials.gov identifier: NCT01144247].
The use of IL-2 in treating brain tumors provides a valuable lesson regarding the challenge of balancing activating the immune system and controlling the toxicity associated with these therapies. It also exemplifies the challenges with translation of preclinical animal models to human therapies. There remains hope that future cytokine therapies, particularly cytokine-secreting cells, could travel beyond the resection cavity, targeting the immunosuppressive microenvironment of invasive and disseminated tumors. Preclinical work also suggests benefit for combinatorial therapy for intratumoral cytokines with systemic checkpoint-inhibitor therapy, however, there is appropriate concern for combinatorial toxicity in humans. 163 The history of limited clinical benefit and significant morbidity and mortality associated with these therapies should require extreme caution with further exploration of immune stimulants in brain tumors.
Future directions
Immunotherapy holds promise despite continued concerns for toxicity. Using the immune system to target and treat cancer may be best chance of increasing survival, while protecting the remaining healthy brain. Further work in multiple domains is needed to optimize therapy, including specifically directing appropriate treatment to appropriate patients, identifying optimal immunologic targets and minimizing off-target immune toxicity.
In the work detailed in this review, there are subsets of patients who respond to immune-based treatments, even if the group as a whole has not yet demonstrated efficacy. This supports the need for a better understanding of host biology to guide appropriate therapies to the patients that may respond. One strategy to address host factors is the ongoing work to describe molecular subtypes of GBM, based on gene expression. 164 Retrospective analysis suggested that the mesenchymal subtype of GBM responded better to an autologous tumor lysate DC vaccination with both greater tumor-infiltrating lymphocytes and increased survival compared with nonvaccinated mesenchymal subtype patients.165,166 Patients with high levels of TGF-β2 also respond more to this therapy. 42 Identifying the molecular factors that affect the interplay of the immune system and the tumor may be the key to matching the therapy to the patient. It is also possible that biomarkers such as PD-L1 expression will guide therapy choices like it currently does in NSCLC, or serum immune response to vaccination will guide continuing therapy. 68 Just as response to EGFRvIII vaccination improved with selection of patients with EGFRvIII tumors, further patient selection will be necessary to target therapy for optimal patient benefit.
Additionally, a number of new approaches are in development, including new checkpoint inhibitors, like signal transducer and activator of transcription 3 (STAT-3), PD-L1 and lymphocyte-activation gene 3 (LAG-3). Newer technologies, such as bispecific T-cell engaging antibodies (BiTES) or CAR-Ts may demonstrate an increased efficacy over the prior generation of treatments if they are better able to elicit a cytotoxic T-cell response.
Given the anatomical and immunologic restrictions of the brain, there have long been concerns that an unchecked immune response in the brain would cause toxicity. Though this concern has lead to a more cautious approach when targeting brain tumors than systemic malignancies, some patients have indeed had tremendous, detrimental CNS inflammation. It may be that off-target autoimmune toxicity is a necessary, but transient consequence of using the immune system to treat malignancy. If that is the case, diligent work is needed to anticipate, monitor and optimize treatment for these toxicities, as preclinical work has not accurately predicted the incidence of these events.
Conclusion
Patients with both primary and metastatic brain tumors have poor overall prognoses. Immunotherapy has revolutionized the treatment of melanoma and other solid tumors and remains a promising strategy for killing infiltrative brain tumors, while protecting the normal brain. The history of immunotherapy in brain cancers serves as a reminder of the risks of treatment in an anatomically and immunologically restricted region. The limited successes and knowledge that have been gained from these studies hold great hope for advancement in the near future.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.