
Abstract
Endocrine therapy is the mainstay of treatment of estrogen-receptor-positive (ER+) breast cancer with an overall survival benefit. However, some adaptive mechanisms in the tumor emerge leading to the development of a resistance to this therapy. A better characterization of this process is needed to overcome this resistance and to develop new tailored therapies. Mechanisms of resistance to hormone therapy result in activation of transduction signal pathways, including the cell cycle regulation with cyclin D/CDK4/6/Rb pathway. The strategy of combined hormone therapy with targeted agents has shown an improvement of progression-free survival (PFS) in several phase II or III trials, including three different classes of drugs: mTOR inhibitors, PI3K and CDK4/6 inhibitors. A recent phase III trial has shown that fulvestrant combined with a CDK 4/6 inhibitor doubles PFS in aromatase inhibitor-pretreated postmenopausal ER+ breast cancer. Other combinations are ongoing to disrupt the interaction between PI3K/AKT/mTOR and cyclin D/CDK4/6/Rb pathways. Despite these successful strategies, reliable and reproducible biomarkers are needed. Tumor genomics are dynamic over time, and blood-based biomarkers such as circulating tumor DNA represent a major hope to elucidate the adaptive mechanisms of endocrine resistance. The optimal combinations and biomarkers to guide this strategy need to be determined.
Introduction
The estrogen receptor (ER) pathway is directly or indirectly activated by estrogen leading to proliferation, differentiation, invasion and cell survival. This pathway is considered an addictive oncogenic pathway in breast cancer cells. In frontline therapy, response rates to anti-estrogenic agents range from 20 to 40%, with a median duration of response of 14 months. In second-line treatment, resistance to anti-estrogenic agents is relatively high as the response rates are less than 10%, with a median progression-free survival of around 4 months.
There is no clear definition of the resistance to hormone therapy (HT). ESO–ESMO guidelines define primary endocrine resistance as: relapse while on the first 2 years of adjuvant endocrine therapy (ET) or progression of disease (PD) within the first 6 months of first-line ET for metastatic breast cancer. Secondary (acquired) resistance is defined as relapse while on adjuvant ET but after the first 2 years, or relapse within 12 months of completing adjuvant ET, or PD 6 months after initiating ET for metastatic breast cancer (MBC). 1
The expression of ER is not sufficient to identify patients who will respond to anti-estrogenic therapy. Several adaptive mechanisms of escape to anti-estrogenic therapy have been identified: an increase in concentration of estrogen in the tumor environment, post-translational modifications, ligand-independent activation of the receptor via ESR1 gene mutation, amplification of the feedback loops mediated by transmembrane growth factor receptors [human epidermal growth factor receptor (HER), fibroblast growth factor receptor (FGF-R), insulin growth factor receptor (IGF-R)], by the RAS/rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase (MAPKinase) pathway and the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway. 2 Downstream, hormone therapy resistance could be characterized by deregulation of the cell cycle involving the cyclin D/CDK4/6/Rb pathway. 3 Several combination strategies with HT have failed in advanced breast cancer such as that combining EGFR inhibitors (gefitinib) and IGFR 1 inhibitors (ganitumumab).4,5 Other combinations with a SRC (rous sarcoma protein) inhibitor (dasatinib), an antiapoptotic inhibitor (bortezomib) or with histone deacetylase inhibitors (HDAC inhibitors) have shown discordant or interesting responses and need more investigations (Table 1).6–8
Trials with endocrine therapy resistance (without PI3K/Akt/mTOR or CDK4/6/Rb pathways inhibition).

Dasa, dasatinib; let, letrozole; exe, exemestane; enti, entinostat; gan, ganitumab; gef, gefitinib; ana, anastrozole; ful, fulvestrant; trast, trastuzumab; lapa, lapatinib; SRC, sarcoma; HDAC, histone deacetylase inhibitors; PFS, progression-free survival.
We hypothesize that addition is more efficient than a substitution strategy for the treatment of endocrine-resistant MBC, as shown in the HER2+ MBC context. 9 We will review randomized phase II and III clinical trials exploiting this strategy.
The proof of concept of adaptive mechanisms with PI3K/Akt/mTOR inhibitors
The PI3K/Akt/mTOR pathway plays a key role in cell signaling, regulating proliferation, survival and differentiation. 10 The PI3K proteins are kinases divided into three classes (I-III) according to their structure and substrate specificity.
Aberrant activation of the PI3K/Akt/mTOR pathway plays a major role in the mechanisms of resistance to HT. It is a prime target for the treatment of ER-positive breast cancers, the aim being to prevent and revert resistance to HT.11–13 The IGF/IGF-1-IRS pathway (insulin receptor substrate 1) induces activation of PI3K and activates mTORC1 with an indirect correlation between PI3K and the mTOR1 effector. Inactivation of PI3K induces inhibition of S6K1 and 4E-BP, and PI3K activation is negatively regulated by the tumor suppressor gene PTEN (phosphatase and tensin counterpart deleted one chromosome ten). The main effector of PI3K is AKT.14,15 This serine/threonine kinase belongs to the family of AGC kinases and exists in three isoforms, Akt-1, 2, 3, encoded by three different genes. Activated AKT induces the activation of the mTOR pathway promoting cell proliferation and inducing inhibition of proapoptotic proteins. MTOR was identified in the yeast Saccharomyces cerevisiae as a therapeutic target of the macrolide antibiotic, rapamycin. 16 It plays a key role in the regulation of critical cell processes such as growth, proliferation, cytoskeleton organization, transcription, protein synthesis, ribosome biogenesis and autophagy17,18 (Figure 1).

PI3K/Akt/mTOR signaling pathway. 18
Hyperactivation of the PI3K/Akt/mTOR pathway will induce tumor adaptation to anti-estrogenic therapy and can be defined by a mutation of PI3K (catalytic domain, or helical), AKT mutation, loss of PTEN function (deletion or loss of expression, epigenetics) or by the regulatory function of proteins TSC1/TSC2 (tuberous sclerosis complex) (deletion–mutation) (Figure 2).
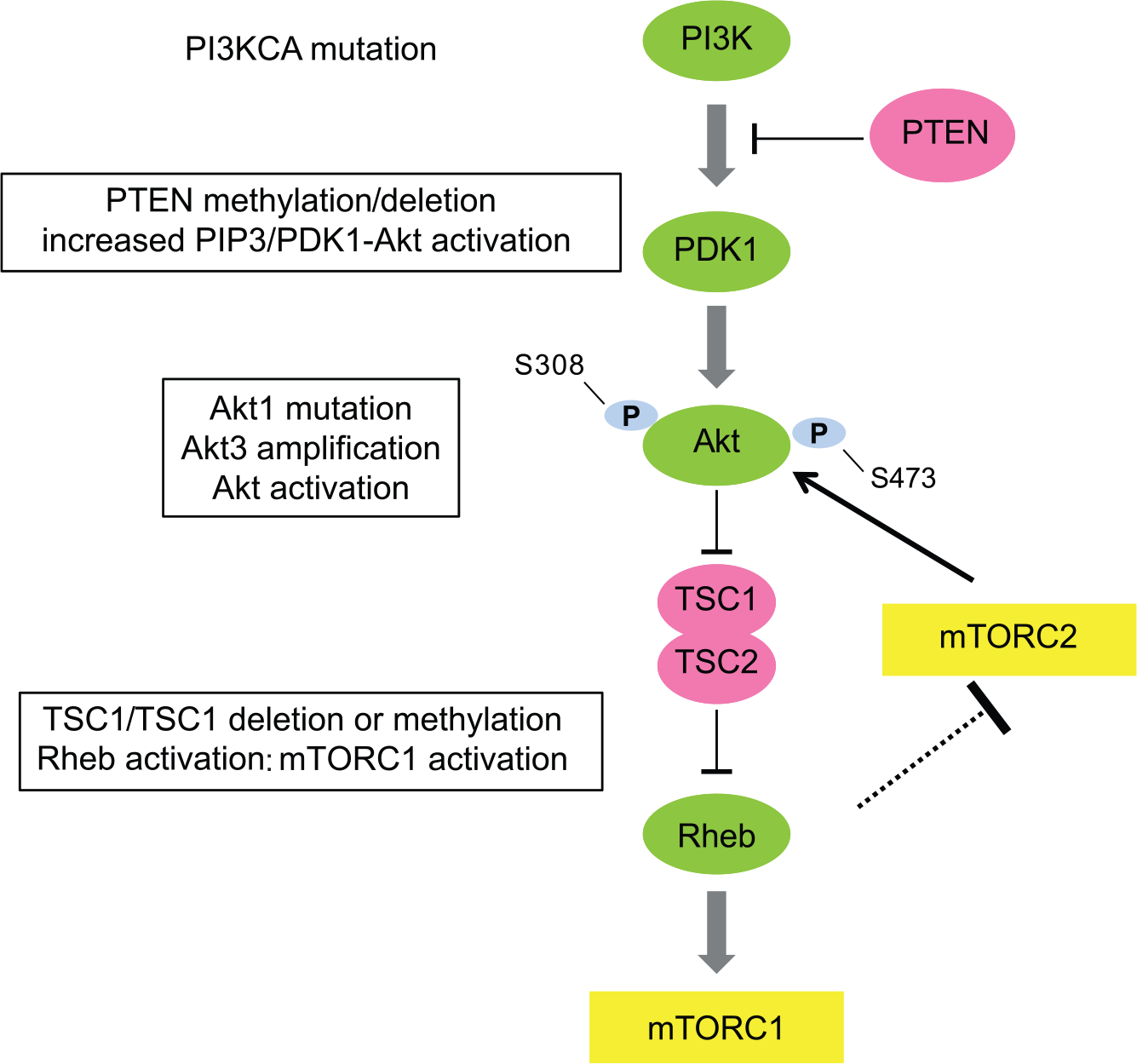
Pathway hyperactivation defined by alterations of the PI3K/AKT/mTOR pathway.
Randomized clinical trials with mTOR inhibitors
Several clinical studies have been conducted in patients with ER + (endocrine receptor) HER2- MBC by combining anti-estrogenic [selective estrogen receptor modulators (SERM), selective estrogen down regulators (SERD)], antiaromatase inhibitors (AI) with agents targeting PI3K/Akt/mTOR such as PI3K inhibitors (panspecific or specific to the subunit 110 α or δ), AKT inhibitors, mTOR inhibitors or inhibitors of both mTOR and PI3K (dual inhibitor).
The
The
What may explain the differences between Horizon, TAMRAD and BOLERO-2?
Unlike BOLERO 2 and TAMRAD, the Horizon study is a first-line setting MBC with only 40% of the patients having received previous adjuvant endocrine therapy. In Horizon, patients didn’t receive any aromatase inhibitor as adjuvant therapy, and likely didn’t have any hyperactivation of the PI3K/Akt/mTOR. MTOR inhibitor might be less effective without an adaptive mechanism as hyperactivation of the PI3K/AKt/mTOR, induced by hormone-therapy resistance.
There is a real heterogeneity of responses to these drugs and patient outcomes, and biomarker analysis may help identify patients who will derive the greatest benefit from everolimus. In an exploratory analysis of BOLERO-2, the benefit of everolimus was maintained regardless of the presence or absence of an alteration in PIK3CA, FGFR1, CCND1 or their respective pathways. However, when the patients were assigned to the subgroups on the basis of mutations in PI3KCA exon 20 or 9, PFS benefit of everolimus appeared to be greater in those with exon 9 mutation [hazard ratio 0.26, 95% CI (0.12–0.54)] than in those with exon 20 mutation [hazard ratio 0.56, CI (0.31–1)]. Moreover, these data suggest that tumor with low chromosomal instability (CIN) might benefit from the addition of everolimus to exemestane, with a median PFS gain of 5.5 months for patients with CIN score below the 75th percentiles [hazard ratio 0.39, CI (0.28–0.54)]. 26
Recently, recurrent mutations have been identified in the estrogen receptor. Chandarlapaty and colleagues evaluated blood samples from 541 of the 724 patients enrolled in BOLERO-2. They detected a D538G ESR1 mutation in samples from 114 (21.1%) patients, a Y537S ESR1 mutation in samples from 72 (13.3%) patients, and double mutations in samples from 30 patients. Median overall survival was 32.1 months for patients with neither a D538G nor Y537S ESR1 mutation, 26 months for those with only a D538G mutation, 20 months for those with only a Y537S mutation, and 15.2 months for those with double mutations. Exploratory analyses showed that adding everolimus to exemestane doubled PFS for patients with any ESR1 mutation and for those with a D538G mutation, and didn’t increase PFS for patients with a Y537S mutation. However, further studies are needed before the validation of these biomarkers. 27
The mechanism of action of everolimus could lead to incomplete inhibition of mTORC1-dependent protein synthesis, limiting its efficacy. 28 Everolimus sets off a negative feedback mechanism leading to increased Akt signaling and treatment resistance. AZD2014, a dual inhibitor of mTORC1 (rapamycin-sensitive) and mTORC2 (rapamycin insensitive) has shown superior activity to everolimus both in hormone-sensitive and -resistant models. 29 A randomized phase II trial (MANTA) [ClinicalTrials.gov identifier: NCT02216786] is ongoing for postmenopausal women with ER+/HER2-negative ABC, hormone refractory, comparing AZD2014 and fulvestrant to fulvestrant alone and to fulvestrant and everolimus.
Randomized clinical trials with PI3K inhibitors
One phase II trial and one phase III trial evaluated a PI3K inhibitor in ER+/HER2-MBC.
Recently, the
The discrepant results regarding the PIK3CA status and the benefit of PI3K inhibitors can probably be explained by the fact that the primary tumor is not suitable for measuring PI3K activation which should probably be analyzed in the metastatic lesions or in circulating DNA at the time of metastatic relapse. In a study of 104 patients, synchronous genetic heterogeneity and changing PI3K mutational status were demonstrated between primary and metastatic breast tumors. 35 The analysis of PI3K mutation status on the circulating tumor DNA (ctDNA) is more feasible, less invasive and more reliable as shown by the unpublished results of the BELLE-2 study. However, prospective well conducted studies are still needed to confirm whether the presence of a PIK3CA mutation in circulating tumor DNA can predict response to this treatment.
CDK 4/6 inhibition in association to AI in a first- or second-line setting?
The mechanisms of resistance to HT often include upregulation with or without activation of signal transduction pathways that involve cell cycle regulation. CDK4/6 phosphorylates and inactivates Rb (retinoblastoma) tumor suppressor proteins, leading to dissociation of E2F transcription factors and transcriptional regulation of genes for G1/S transition and cell cycle progression. Mitogenic signals or growth factor receptor signaling pathways converge on the cyclin D/CDK4 or CDK6 pathways, like ER and PI3K/Akt/mTOR. The activation leading to Rb phosphorylation is associated with resistance to endocrine therapy.3,36 Rb dysfunction is associated with luminal B-type breast cancer and is predictive of a poor response to endocrine therapies. CDK 4/6 inhibitors reverse the endocrine resistance in preclinical studies. 37
However, fulvestrant could be a new option in a first-line setting. Fulvestrant is a selective estrogen-receptor degrader that targets the function of the hormone receptor currently used in second-line after AI.
40
A randomized open label phase II trial, the
Ribociclib has been evaluated in the
The
Regardless the CDK 4/6 inhibitor, biomarkers are needed to optimize endocrine therapy and the levels of estrogen (ER) and progesterone receptor (PgR) could be one. Low proliferation with a low Ki67 level and high eostrogen and progesterone receptor expression (progesterone receptor) are probably predictive of response to endocrine therapy in neoadjuvant BC. 47 Low PgR and higher Ki67 expression seem to be associated with poor prognosis and help to strengthen HT. Indeed, in the TEXT and SOFT trials, Regan and colleagues showed that the combination of exemestane + ovarian function suppression (OFS) is more beneficial than tamoxifen (tam) alone or tam + OFS in adjuvant endocrine therapy for women with poor prognostic features. 48 In patients with newly metastatic disease or disease recurring after adjuvant tamoxifen, negative or low-level ER could be a predictive factor of response to gefitinib (EGFR inhibitor) and tamoxifen. 49 No biomarkers have been found to predict the response of CDK4/6 inhibitors. In the PALOMA-2 trial, biomarker analyses on cell-cycle-related genes using immunohistochemistry for ER, Rb, p16, cyclin D1, and Ki-67 revealed no additional markers with sensitivity to palbociclib + letrozole beyond ER+. 50 A recent phase II study evaluated if short-term preoperative palbociclib treatment is associated with decreased proliferation and early biomarker changes in patients with early breast cancer. Palbociclib decreases Ki67 and is dependent on molecular subtypes; it is not effective on HER2+ and triple-negative breast cancer, and is correlated with changes in pRB. Additional analyses are ongoing (CCND1 amplification, pAKT, pER, PIK3CA, AKT1). 51
Given these remarkable results, acquired resistance to CDK4/6 inhibitors will be an emerging clinical challenge.
Novel combinations
The interaction between the PI3K/AKT/mTOR and cyclin D–CDK4/6–INK4–Rb pathways is thought to play a critical role in ER-driven breast cancer and preclinical ER+ breast cancer models. Hyperactivation of PI3K/Akt/mTOR might be an adaptive mechanism leading to endocrine therapy and CDK4/6 inhibitor resistance, and combination strategies are widely evaluated. The combination of ribociclib, a CDK4/6 inhibitor (LEE011; LEE), alpelisib, an alpha isoform of class I PI3K inhibitor (BYL719; BYL), and letrozole (LET) has recently shown enhanced activity versus each agent alone. 52 A phase Ib combination of LET, LEE and BYL has shown an acceptable safety profile and demonstrates preliminary clinical activity in heavily pretreated patients with ER+/HER2- ABC. A total of 15 patients discontinued treatment: 7 (19%) due to progression of disease (PD) and 8 (22%) due to AEs. The most frequent study drug-related AEs (all grades >35%) were: nausea (all grades, 44%; G3/4, 6%), hyperglycemia (44%; 17%), neutropenia (42%; 22%), and fatigue (36%; 11%). Among 27 evaluable patients, 2 (7%) had a partial response (PR), 4 (15%) had unconfirmed partial response, 6 (22%) had stable disease (SD), 6 (22%) had non-CR (complete response), non-PD, and 5 (19%) had PD as best overall response. Inhibition of the three pathways provides sustained downregulation of Ki67, potentially preventing a feedback mechanism and hence delaying progression through therapy. Future randomized studies will compare LET + LEE or BYL with LET + LEE + BYL. 53
In summary, practice-changing trials have been successfully conducted combining HT with targeted therapies, disrupting the adaptive mechanisms of resistance to HT. This illustrated the successful concept of adding therapies. Fulvestrant as monotherapy is also an option, but recent trials have shown that PFS benefits can be achieved from it being combined with an inhibitor of the cyclin D–CDK4/6–INK4–Rb pathways.42,43 Fulvestrant, in combination with a PI3K inhibitor could be another option, but trials are still ongoing. Everolimus + exemestane is also an option in a second-line setting in postmenopausal women with ER+ cancer with prior exposure to nonsteroidal anti-inflammatories (letrozole or anastrozole), despite the absence of OS improvement (Figure 3). 22 New and robust biomarkers are needed to define what is the best combination and the best sequence of treatment for hormone refractory MBC. To monitor the dynamics of tumor genomics over time, reliable and reproducible biomarkers for given patients are urgently needed. And ctDNA seems to be a useful one. There is a real interest in integrating liquid biopsies in prospective combination trials, but the sensibility and reproducibility of these tests are still being evaluated.

Resistance to endocrine therapy and adaptive mechanism in advanced breast cancer.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.