
Abstract
The chronic, progressive clinical characteristics of many adult solid tumor malignancies suggest that a more effective therapeutic approach to cancer management may require long-term intervention using nontoxic systemic agents that block critical components of abnormal tumor physiology. Two highly promising systemic targets common to the development, progression and recurrence of many common cancers are dysregulated inflammatory and oxidation/reduction (redox) pathways. Compelling clinical data support the use of anti-inflammatory and antioxidant agents as a therapeutic modality for long-term use in patients diagnosed with several common cancers, including colon cancer and breast cancer. The therapeutic paradigm presented in this paper is the product of a synthesis of what is currently understood about the biological effects of inflammation and oxidative stress that contribute to tumorigenesis, disease progression and recurrence as well as results obtained from research on the use of prophylactics with anti-inflammatory or antioxidant properties in cancer prevention and treatment.
Introduction
Most current standard of care therapeutic approaches to systemic cancer involve acute, short-term treatment regimens (weeks to months) of cytotoxic chemotherapy drugs in combined, dose-dense treatment protocols. The early success in the application of this therapeutic approach to the treatment of childhood acute lymphoblastic leukemia (ALL) became the model for the further development of cytotoxic chemotherapy drugs for the treatment of many types of cancer. Most cytotoxic chemotherapy drugs in widespread use today, including paclitaxel, fluorouracil, gemcitabine, fludarabine, BCNU, carboplatin, cytosine arabinoside, pentastatin, hydroxyurea, topotecan, mitoxantrone and others are either S-phase, M-phase or phase nonspecific cell cycle inhibitors. In the field of cancer genetics, the discovery of the significance of mutated cellular oncogenes and tumor suppressor genes as primary causes of malignant transformation not only provided a theoretical rationale for the use of traditional chemotherapy drugs such as methotrexate and paclitaxel that block cell cycle progression, but also laid the groundwork for a new era of cancer medicine based on the development of monoclonal antibodies and small molecule therapeutics designed to block the activities of runaway genes directly implicated in cancer causation. The therapeutic success of imatinib mesylate (also known as Gleevec Novartis), an inhibitor of the oncogenic abl US Food and drug Administration gene activated by reciprocal transformation in chronic myeloid leukemia (CML), showed that gene targeted anticancer drugs can have a dramatic and profound therapeutic efficacy in patients with CML or gastrointestinal stromal tumors whose cancers contain this mutation.
The Gleevec success story opened the door to the era of personalized cancer medicine that is based on the concept that the identification of specific genetic lesions in individual patient tumors will facilitate the appropriate use of gene-targeted drugs tailored to eradicate a malignancy based on its assessed genetic profile. This gene-targeted therapeutic approach led to the US Food and Drug Administration (FDA) approval of trastuzumab (Herceptin (Genentech/Roche)), a targeted monoclonal antibody, as a treatment for patients with breast cancer who test positive for its genetic target, the amplified human epidermal growth factor receptor 2 (HER2)/neu oncogene. Likewise, gene-targeted therapeutics cetuximab and panitumumab were approved for use in patients with colon cancer containing the oncogenic ras mutation. Recently, the FDA approved afatinib (Gilotrif, Boehringer Ingelheim Pharmaceuticals) a monoclonal antibody that targets mutations in the epidermal growth factor receptor gene (EGFR) that occur in about 10% of patients with non-small cell lung carcinoma. Afatinib was approved in conjunction with a genetic test for the presence of this mutation as a companion tool for the identification and treatment of patients with this type of lung cancer. Approval was based on clinical trial results showing that patients who received afatinib in place of standard chemotherapy (pemetrexed and cisplatin) had a delay in disease progression of approximately 4 months, although there was no difference in overall survival between the two groups of patients. Also, in 2013 the FDA approved erlotinib (Tarceva Genentech/Roche) for the treatment of patients with non-small cell lung cancer and a companion genetic test for the EGFR mutation. Please see Table 1 for a more complete list of FDA-approved gene-targeted cancer therapeutics.
List of recently approved gene-targeted drugs by the US Food and Drug Administration (FDA).

Source: My Cancer Genome (managed by the Vanderbilt-Ingram Cancer Center).
ALL, acute lymphoblastic leukemia; CL, chronic myeloid leukemia; EGFR, epidermal growth factor receptor; GI, gastrointestinal; HER, human epidermal growth factor receptor; VEGFR, vascular epidermal growth factor receptor; SEER, surveillance, epidemiology and end results program; CDC, centers for disease control.
The efficacy of gene-targeted therapeutics in the treatment of select cancer types is undisputed; nevertheless, the success rate of traditional cytotoxic chemotherapy in producing long-term patient disease-free survival is unpredictable, and in many cancers, unsatisfactory (see Figure 1). Thus, despite many recent advances in clinical drug development, some of the most common cancers continue to show inconsistent and incomplete therapeutic responses to the current standard of care: combined chemotherapy approaches using conventional cytotoxic cell cycle inhibitors with or without additional gene-targeted therapeutics. Consequently, for many patients with systemic disease, current therapeutic approaches are successful only in delaying the time to disease progression rather than affecting long-term survival rates. Once therapy is discontinued, the local and systemic conditions that produced the original tumor may set the stage for disease progression or recurrence. Moreover, recurrent disease is frequently aggressive and therapy resistant, further complicating the clinical management of patients with cancer. Further advances in achieving long-term disease-free survival rates for many types of cancer may require additional therapeutic modalities that systemically target critical physiological processes responsible for disease recurrence (Crawford, 2013). Moreover, to achieve this therapeutic goal, it may be necessary to implement long-term nontoxic chemotherapy regimens in a maintenance therapy modality to provide ongoing systemic inhibition of disease progression or recurrence.

Age-adjusted mortality rates for all cancers combined, US SEER data [Howlader et al. 2011]. SEER, surveillance, epidemiology and end results program; CDC, centers for disease control.
The concept of long-term maintenance therapy approaches to prevent disease recurrence is supported by longitudinal studies on the long term use of tamoxifen and aromatase inhibitors in the management of estrogen receptor (ER)-positive breast cancer [Early Breast Cancer Trialists’ Collaborative Group et al. 2011]. The consensus of these clinical studies was that, despite the occurrence of side effects, this approach was successful in preventing disease recurrence by targeting growth factor and hormonally activated pathways important to sustaining tumor progression and disease recurrence.
Oxidative stress/inflammation: primary role in disease progression
The successful application of long-term maintenance therapeutic approaches to cancer, of course, will necessitate the identification of critical local and systemic factors responsible for disease progression and recurrence. Physiological studies suggest that genetic mutations that affect cell proliferation rates may not be the sole determinants of cancer progression, a process that may be affected significantly by immune system dysfunctions, particularly those that involve generalized immunosuppression and the activation of proinflammatory cell pathways. Even the process of metastasis itself, the hallmark of lethal cancer progression, does not appear to be the direct result of primary genetic mutations, but rather the end result of complex intersecting ‘chaotic’ pathways that may result in the progression of a localized tumor to systemic disease. As such, these pathophysiological phenomena represent the accumulation and elaboration of chronic disease mechanisms that, in many cases, cannot be approached using acute therapy regimens designed solely for short-term cytotoxic effects.
Extensive evidence described here and elsewhere supports the concept that dysregulated inflammatory and redox pathways in tumor cells and their stromal environment play an essential role in tumor genesis, invasion and systemic spread. For example, many cancers, especially solid tumors such as colon cancer, pancreatic cancer, breast cancer, liver cancer, stomach cancer, cervical cancer, prostate cancer, lung cancer and others are preceded by infection or inflammation within the organ in which the cancer arises; moreover, inflammatory pathways are constitutively active in most cancers (see Figure 2) [Rolland et al. 1980; Kune et al. 1988; Coussens and Werb, 2002; Farrow and Evers, 2002; Pai et al. 2002; MacArthur et al. 2004; Nelson et al. 2004; Philip et al. 2004; Sheng et al. 2001; Thun et al. 2002, 2012]. Tumor pathways associated with survival, proliferation, invasion and metastasis are all important activators of nuclear factor κB (NFκB) and signal transducer and activator of transcription 3 (STAT-3), major transcription factors that regulate inflammatory pathways activated by endotoxins, carcinogens, radiation, chemotherapy, hyperglycemia, tumor promoters, inflammatory cytokines [e.g. tumor necrosis factor, cyclooxygenase 2 (COX-2), interleukin 1] and growth factors such as EGF [e.g. Rolland et al. 1980; Philip et al. 2004; Itzkowitz and Yio, 2004]. In addition, almost all infectious agents associated with cancer cause inflammation, including human papilloma virus, human herpes virus, hepatitis B virus and hepatitis C virus [Peek and Blaser, 2002; Itzkowitz and Yio, 2004]. Chemotherapy and radiation also activate inflammatory pathways, which subsequently contribute to acquired radiation and chemo resistance. Based on a large volume of research data, including the aforementioned and other studies, one may conclude that most gene products associated with inflammation can contribute to cancer development as well as progression, survival, proliferation, invasion, angiogenesis and metastasis, all of which are regulated by NFκB and STAT-3.

Primary relationships among carcinogens, activated reactive oxygen species (ROS) and inflammatory responses mediated by nuclear factor κB (NFκB) [Singh et al. 2011]. IκB, inhibitor of κB.
Likewise, reactive oxygen species (ROS) appear to play a critical role in cancer (see Figure 3). ROS are produced as a byproduct of oxygen metabolism as a consequence of the incomplete reduction of oxygen to produce the superoxide anion O2–, a highly reactive moiety that is the precursor of hydroxide (.OH), hydrogen peroxide (H2O2) and peroxynitrite (OONO–). Mitochondrial production of ROS as a byproduct of oxidative respiration is enhanced by the aging process and by exposure to carcinogens, radiation and other environmental toxins that produce elevated levels of intracellular ROS [Kovacic and Osuna, 2000; Wardman, 2001]. The overall result is elevated levels of oxidative stress. Oxidative stress may also occur as a result of reduced enzymatic antioxidant activity as well as growth factor receptor interactions, cytokines, shear stress, chemotherapy, ionizing radiation and aging. In turn, ROS may induce stress responses to maintain energy metabolism, but at high levels this causes broad-spectrum oxidative damage that may elicit apoptosis by inducing membrane permeability changes in mitochondria resulting in the release of cytochrome C. Oxidative damage produces deletions and duplications in mitochondrial DNA, a process that increases with age in many human tissues, further contributing to mitochondrial dysfunction [Miqueland Bertoni-Freddari, 2000]. Moreover, mitochondrial DNA is much more sensitive to mutagenesis by ROS than nuclear DNA, which may contribute to impairments of mitochondrial function under conditions of elevated intracellular ROS.

Inflammation, oxidative stress and tumor physiology: a ‘perfect storm’ for tumor spread, metastasis and recurrence. Reactive oxygen species (ROS)/hypoxia damage mitochondria and results in further ROS production and the glycolytic switch to activate inflammatory pathways. Hypoxia inducible factor 1α (HIF-1α) drives survival and metastatic pathways, involving nuclear factor κB (NFκB), STAT-1 and autophagy. Integrin-associated focal adhesion complexes also enhance ROS to destabilize cadherin-associated adherens junctions [Maiti, 2012].TNF, tumor necrosis factor. APEX-1, multi-functional DNA repair enzyme; ARHGEF-6, rho guanine nucleotide exchange factor; CAT, catalase; CDK-6, cyclin dependent kinase; CYR61, cysteine-rich angiogenesis inducer; GPX-1, glutathione peroxidase; GSR, glutathione reductase; GSTM-1, glutathione S transferase; GSTP1, glutathione S transferase P1; HSP 1A,1B, heat shock protein; KEAP-1, kelch-like ECH associated protein; MAPK-8; mMAP kinase; MIR-154,-648,-1321, microRNA: NRF2, nuclear factor;NFE2L2, nuclear factor erythroid derived-like; PAK2, p21 protein activated kinase; PI3K, phosphatidyl inositol-3-kinase; PRDX-4, peroxyredoxin; SOD-2, superoxide dismutase; TNF, tumor necrosis factor; TP53, tumor suppressor protein; TXN, thioredoxin.
Thus, there appears to be a cyclical interaction between mitochondrial respiration, which is an important source of ROS due to electron leakage from the respiratory chain, and the destructive effects of excessive ROS production on mitochondrial function. Furthermore, decreased mitochondrial activity may act as a trigger of the glycolytic switch characteristic of tumor cell metabolism. Thus, mitochondrial damage in cancer cells may in part contribute to enhanced ROS production to initiate a cycle of events in surrounding stromal tissue that ultimately restores energy balance via mitochondrial adenosine triphosphate production in the tumor.
Research by Ishikarwa and colleagues provided evidence that ROS-generating mitochondrial DNA mutations may regulate tumor cell metastasis [Ishikarwa et al. 2008]. This research showed positive correlations between mitochondrial dysfunction, increased ROS and metastatic spread. Cytoplasmic hybrid technology (cybrid) was used to transfer mitochondrial DNA from aggressive mouse tumors to nonaggressive tumors and vice versa. High levels of tumor metastasis were observed to occur only in cells containing mitochondrial DNA from aggressive tumors. These studies also showed that mutations in the NADH gene were linked to high metastatic potential. These mutations produced a defect in respiratory I complex activity and a concomitant increase in ROS. Moreover, it was observed that pretreatment with ROS scavengers abrogated the metastatic potential of the mutated mitochondrial DNA. Nuclear genes affected by the mitochondrial mutations that promote metastasis include vascular endothelial growth factor, hypoxia inducible factor 1α (HIF-1α) and myeloid cell leukemia sequence 1.
Important recent research studies on the relationship between tumor cells and surrounding stromal fibroblasts, termed cancer-associated fibroblasts (CAFs), in breast and other cancers have shown that H2O2 produced by oxidative stress in tumor cells may be taken up by surrounding stromal tissue to initiate autophagy and mitophagy of the nontumor tissue [Martinez-Outschoorn et al. 2010a, 2010b, 2010c]. The resulting tissue destruction results in the production of nutrients and other metabolites that are subsequently consumed by the tumor cells to promote tumor growth. Moreover, elevated levels of H2O2 resulting from oxidative stress activate autophagy in CAFs that, in turn, results in the loss of membrane-bound caveolin 1 (cav-1), an important negative regulator of signal transduction pathways. Preclinical research has shown that cav-1 is degraded via autophagy in response to oxidative stress in human fibroblasts cocultured with breast cancer cells [Bonuccelli et al. 2010a, 2010b]. Decreased levels of cav-1 in tumor stromal tissue correlate with poor prognosis in breast cancer, prostate cancer and melanoma [Witkiewicz et al. 2006]. Loss of cav-1 is associated with tissue fibrosis, extracellular membrane (ECM) remodeling and metabolic reprogramming in CAFs, all of which contribute to disease progression and metastasis. Moreover, the loss of stromal cav-1 has been linked to a ‘reverse Warburg effect’, resulting in autophagy and mitophagy in tumor-associated fibroblasts that provide energy-producing ketones and lactate to tumor cells in the microenvironment that support tumor growth [Bonuccelli et al. 2010a, 210b; Lisanti et al. 2010]. Furthermore, these studies suggested that breast cancer cells induce the expression of monocarboxylate transporter shuttle (MCT4), the major intercellular lactate transporter in glycolytic cells, in tumor stromal tissue. The levels of stromal MCT4 and cav-1 are inversely related in stromal tissue in patients with breast cancer. Low levels of stromal cav-1 in conjunction with high levels of MCT4 are biomarkers for poor prognosis in women with many types of breast cancer, including ER positive, progesterone receptor positive, HER2/neu positive and triple negative breast cancer (TNB) [Mercier et al. 2008; Di Vizio et al. 2009; Witkiewicz et al. 2009a, 2009b, 2010]. For example, in patients with TNB, high levels of stromal cav-1 correlate with survival rates of over 75% in 12 years. In contrast, patients with TNB with very low levels of stromal cav-1 have a 5-year survival rate of less than 10% [Finak et al. 2008; Sloan et al. 2009]. Moreover, loss of stromal cav-1 is an accurate biomarker for progression to invasive disease in women with ductal carcinoma in situ. Moreover, MCT4 induction, associated with activated stromal recruitment by breast cancer cells, can be blocked by antioxidants such as N-acetyl-cysteine, quercetin and metformin, as well as chloroquine, which inhibits autophagy, all of which have been shown to prevent loss of stromal cav-1 in tissue coculture systems [Trimmer et al. 2011].
Three-dimensional tumor structure: biophysical link to oxidative stress and inflammation
Solid tumor formation involves an abrogation of critical biophysical parameters that regulate organ and tissue homeostasis (see Figure 4). Abnormal tumor vasculature is a primary cause of hypoxia, intratumoral acidity and increased interstitial fluid pressure of the tumor microenvironment [Boucher et al. 1990]. Normal PO2 oxygen ranges between 10 and 80 mmHg; many solid tumors contain PO2 at less than 5 mmHg. The resulting activation of HIF-1α is associated with solid tumor progression and has been shown to drive many of the survival and metastatic pathways that are characteristic of advanced disease, including NFκB activation and autophagy. Moreover, the hypoxic tumor microenvironment is a major cause of tumor reliance on glycolysis for energy production. Increased glycolysis results in elevated tumor acidity compared with normal tissues as a result of lactate accumulation.
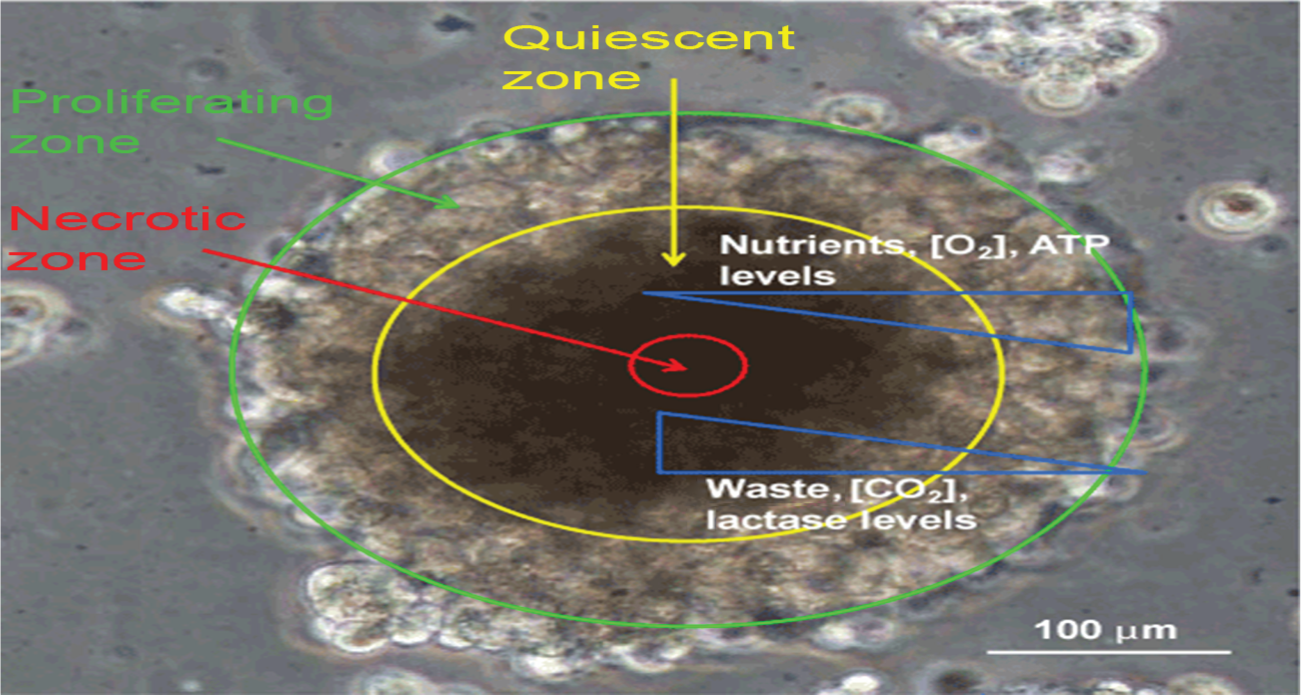
Abnormal tumor microenvironment results from structural parameters generated in the process of solid tumor formation [Chandrasekaran and King, 2012].ATP, adenosine triphosphate.
Oxidative stress is implicated in cell-to-cell and cell-to-stroma interactions that play an important role in three-dimensional tumor growth. Among the morphological effects of oxidative stress are enhanced formation of focal adhesions by integrin matrix complexes and the breakdown of adherens junctions that mediate cell-to-cell contacts by cadherins. Research has shown that integrin–ECM coupling induces changes in mitochondrial redox metabolism to induce elevated levels of intracellular ROS to activate NAD(P)H oxidase 5-lysooxygenase (5-LOX) and COX-2 [Gupta et al. 2012]. Moreover, inside-out ROS signals induce conformational changes in integrins that activate their ECM-binding activities. In this positive feedback loop, integrin-mediated formation of focal adhesion complexes further enhances ROS signaling mechanisms. The effects appear to be mediated by GTPase Rac-1 [Lisanti et al. 2010]. The formation of focal adhesion complexes activates proinflammatory pathways regulated by NFκB. In contrast, contact inhibition of cell growth is associated with low levels of ROS that inhibit growth factor receptor transduction pathways necessary for cell cycle progression. It is possible that at high levels of intracellular ROS, this cell-to-cell contact inhibition of cell proliferation may be abrogated.
Synergistic effects of inflammation and ROS on tumor physiology: rationale for a combined therapeutic modality for disease progression and recurrence
Inflammation and oxidative stress comprise highly interrelated biochemical processes directly implicated in cancer development, progression and systemic spread. Both inflammation and ROS activate NFκB and other transcription factors that drive tumor progression. ROS production is an important component of inflammation during the normal process of wound healing and also in the dysregulated inflammatory pathways activated by oncogenesis. Specifically, elevated intracellular H2O2 activates critical enzymes involved in arachidonic acid metabolism, including phospholipase A2, LOX and COX-2. Overproduction of ROS by damaged mitochondria also activates inflammatory pathways linked to cancer [Miqueland Bertoni-Freddari, 2000; Lisanti et al. 2010; Martinez-Outschoorn et al. 2010c; Pavlides et al. 2010b].
In addition to mitochondrial damage, oxidative stress in tumor cells results from the dysregulated activity of NAD(P)H oxidases, xanthine oxidases, cytochrome P450 mono-oxygenase, uncoupled nitrogen oxide synthase, LOX and COX(s). The latter comprise critical components of arachidonic acid metabolism involved in inflammatory responses that generate reactive lipid peroxides, providing a direct link between cellular redox levels and inflammatory pathway activation. Moreover, NFκB pathway activation, a critical stress activated component of cellular metabolism, occurs as a consequence of inflammation/oxidative stress to produce a ‘perfect storm’ of tumor activity that ultimately drives tumor spread, metastasis and systemic disease (see Figure 3).
Additional evidence for the interrelationship between antioxidant and anti-inflammatory pathways is indicated by the fact that the pharmacological properties of antioxidants and anti-inflammatory agents overlap. Antioxidant natural products with demonstrated anticancer activity that also block inflammation via NFκB pathway inhibition include curcumin, resveratrol, ursolic acid and others [Huang et al. 1994; Jang et al. 1997]. For example, in human clinical trials, curcumin was shown to downregulate NFκB and STAT-3 and is thought to have potential for the prevention or treatment of pancreatic cancer, familial adenomatous polyposis, inflammatory bowel disease, durable bowel disease and other proinflammatory diseases [Gupta et al. 2012]. In addition, research studies have suggested that many cancer preventive agents with antioxidant properties mediate their effects through inhibition of NFκB and STAT-3, critical regulators of proinflammatory pathways in the cell [Baek and Eling, 2006]. For example, the anti-inflammatory effects of vitamin D include inhibition of prostaglandin (PG) synthesis. High levels of COX-2 correlated with low levels of the vitamin D receptor expression in many breast tumors. In vitro studies have shown that vitamin D uptake results in decreased COX-2 levels and the induction of PG dehydrogenase in association with decreased PG receptor gene expression [Fleet et al. 2012].
Clinical evidence of chemopreventive effects of antioxidant/anti-inflammatory agents: potential long-term therapeutic applications
Long-term maintenance therapy protocols that target inflammatory pr oxidative stress pathways implicated in disease progression may be an appropriate choice to initiate clinical studies on the potential benefits of this novel preventive/therapeutic approach, as these agents lack the immune-suppressing systemic cytotoxicity of conventional chemotherapy drugs, which would prohibit their use in long-term disease management. However, research clinical trials designed to evaluate the effects of antioxidant and anti-inflammatory agents have not yielded conclusive findings on their potential efficacious use in the prevention or treatment of cancer.
Much recent attention has focused on the long-term use of anti-inflammatory drugs such as aspirin as cancer preventives. The association between chronic inflammation and inflammatory pathway activation in the genesis of cancer is supported by clinical research studies that provide evidence for the preventive aspects of long-term aspirin use in the development of some common malignancies, including breast, colon cancer, bladder cancer, melanoma and other cancers [Anderson et al. 2002; Castelao et al. 2000; Pereg and Lishner, 2005; Sharpe et al. 2000; Thun et al. 2002; Ulrich et al. 2006; Annemijn et al. 2012] (see Table 2). This preventive effect has also been documented in individuals at high risk for developing colon cancer due to inherited genetic mutations [Burn et al. 2011; Chan and Lippman, 2011; Ulrich et al. 2006].
Combined meta-analysis of clinical trial data from 51 studies on the anticancer effects of aspirin [Langley, 2013].

CI, confidence interval.
Aspirin consumption was shown to prevent the development of precancerous colon adenomas [Cole et al. 2009] and to improve survival odds for patients with colon cancer [Bastiaannet et al. 2012]. Longitudinal studies have shown that long-term daily aspirin use for at least 10 years reduces the risk of developing colon cancer and other common cancers. These clinical data indicated that low-dose aspirin use for at least 3 years may reduce the risk of cancer incidence by about 25% and the risk of dying from cancer by about 15%. The statistic increases from 25% to 37% for those who take aspirin for longer than 5 years [Rothwell et al. 2010, 2011, 2012 a, 2012b].
Clinical results also suggest that regular aspirin use may help prevent the spread or metastasis of cancer to other organs [Annemijn et al. 2012; Rothwell et al. 2012] (see Table 3). These data suggested that long-term daily aspirin use reduced the proportion of cancers that spread systemically by 48%. Moreover, use of anti-inflammatory drugs reduced the risk of being diagnosed with a solid cancer that had already spread by 31%. For patients initially diagnosed with a local cancer, the risk of later metastasis was reduced by 55% by daily high-dose aspirin usage. The study authors suggested that at least part of this preventive effect may be linked to the effects of aspirin on platelets. Moreover, the authors suggested that theirs was the first study to show that any drug could reduce metastasis as a specific drug-induced effect. Suppression of the NFκB regulator, inhibitor of κB kinase β and STAT-3 have been shown to block tumor proliferation and invasion [Pereg and Lishner, 2005]. Moreover, research studies have shown that transcriptional silencing of COX-2 results in suppression of metastasis in TNB [Stasinopoulos et al. 2013].
Summary of clinical trial data on aspirin use and mortality, overall survival and recurrence rates in diverse cancer types [Langley, 2013].
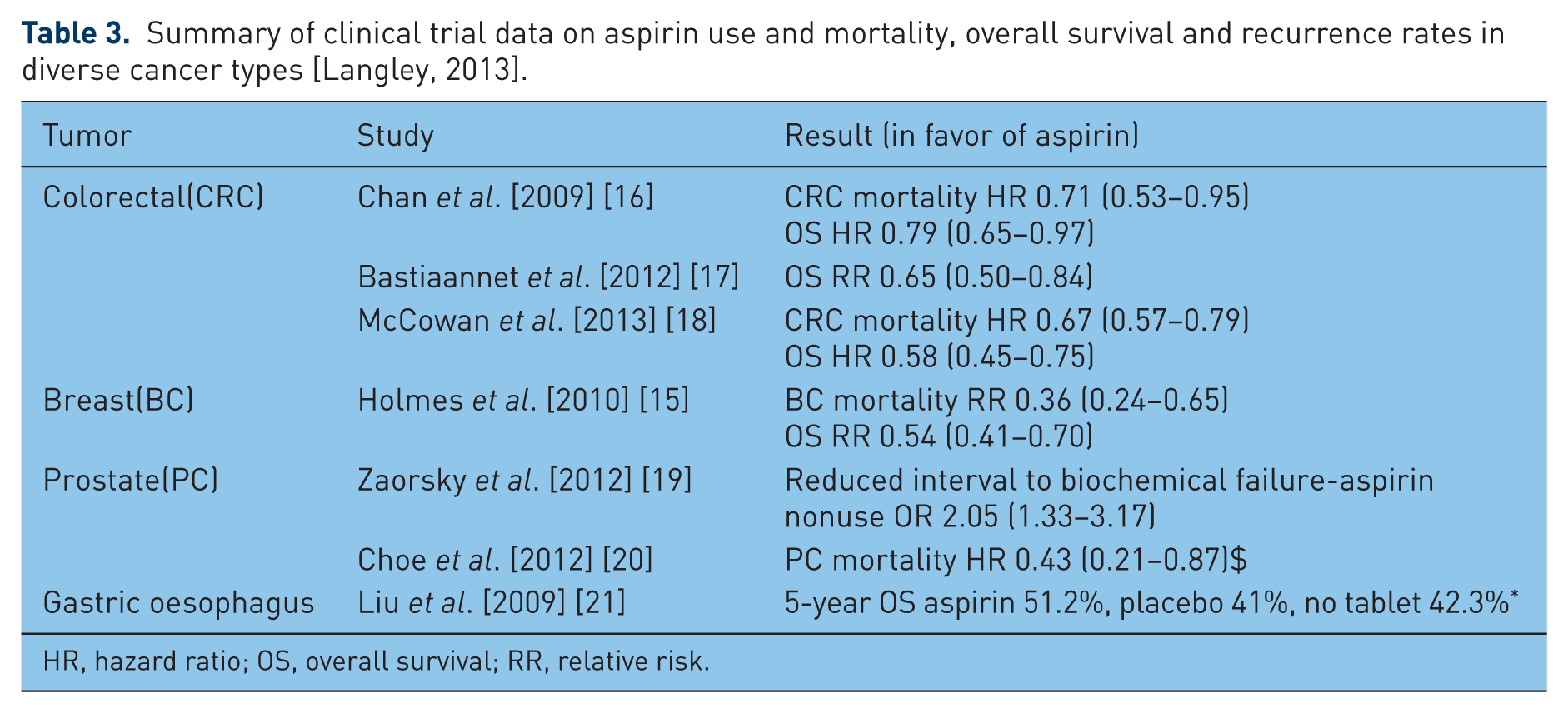
HR, hazard ratio; OS, overall survival; RR, relative risk.
Contradictory research clinical trial findings on the efficacy of nonsteroidal anti-inflammatory drugs in cancer prevention and treatment were reported by Higgins and colleagues as part of the North Central Cancer Treatment Group adjuvant trial. This trial compared the effect of exemestane and anastrozole on breast cancer risk and recurrence and also studied the effects of low-dose aspirin (81 mg daily) and celecoxib on disease parameters [Higgins et al. 2012]. Celecoxib did not positively affect disease parameters and aspirin use was associated with poorer clinical outcome than observed in the control group. The authors noted that a lack of randomization of this portion of the trial prevented a conclusive determination of anti-inflammatory use and prognostic outcome.
The potential preventive and therapeutic applications of antioxidants are more controversial. Both negative and positive effects of diverse compounds with antioxidant activity have been reported; a review of recent clinical data suggests the need for more focused assessments of agents with targeted intracellular effects on specific redox pathways implicated in oncogenesis [Brown and Wilson, 2004; Pouysségur et al. 2006; DeLorenze et al. 2010; Thomasset et al. 2007]. A case in point involves a systematic meta-analysis of 68 clinical trials involving over 250,000 participants on the effects of vitamin supplements β carotene, vitamin A and vitamin E that, strikingly, concluded that their consumption did not increase longevity, but instead produced statistically significant increases in mortality [Bjelakovic et al. 2007, 2012]. Selenium supplementation, in contrast, was found to be associated with slightly more positive effects on longevity. On the surface, the overall conclusions of this large-scale meta-analysis suggest that antioxidant use may be contraindicated as a cancer preventive or therapeutic. A closer inspection of the data, however, suggests that this interpretation of the clinical data may paint a picture with a broad stroke that needs to be refined. The antioxidants evaluated in these clinical trials were synthetic vitamins, rather than natural products; moreover, there is some evidence that β carotene may actually function as a cocarcinogen, eliciting both pro- and antioxidant effects. A distinction could also be made among individual antioxidant supplements with respect to differential effects on longevity; for example, selenium showed a modest benefit.
It is, therefore, critical to evaluate the potential anticancer effects of antioxidants on an individual basis, rather than as a homogeneous group as each chemical exerts different and somewhat specialized effects on cell physiology. Their biological properties are not limited to their antioxidant effects, and other biological properties may contribute to adverse effects on cell physiology, which is especially relevant since this study included many trials in which the dosing regimens were generally high without respect to the potential adverse effects of high-dose supplementation. Specifically, the dosing regimens of antioxidants may need more rigorous calculation as ROS such as H2O2 have important cellular functions; therefore, it is possible that high-dose supplementation may have negative effects on oxidative metabolism. A case in point involves the anticancer effects of the nutriceutical curcumin. Several studies have suggested that, at low concentrations, curcumin displays antioxidant effects, whereas at high concentrations it may act as a pro-oxidant [Gupta et al. 2012]. The ambiguous conclusions reached in these clinical trial analyses suggest the need to evaluate natural phytochemicals as well as enzyme-targeted antioxidants with more precise mechanisms of action.
Important research studies on vitamin D suggest that this vitamin, which exerts pleiotropic effects on systemic and cellular physiology, displays a powerful protective effect against breast cancer, prostate, melanoma and other cancers [Hatse et al. 2012]. These clinical studies have shown that high levels of serum vitamin D at the time of breast cancer diagnosis are associated with smaller tumor size and higher overall survival rates in postmenopausal women (the protective effect was not observed in premenopausal women with breast cancer). Vitamin D is a critical regulator of gene expression that affects the transcription of many genes implicated in growth control; therefore, its protective effect is generalized and cannot be ascribed to a single mechanism. That said, vitamin D plays an important role in modulating inflammatory and oxidative stress pathways that likely contribute to its anticancer effects (for a detailed review, see Fleet and colleagues) [Fleet et al. 2012]. Prominent among these is oxidative regulation by H2O2 of integrin–ECM focal adhesion complexes and cell-to-cell cadherin-associated adherens junctions, both implicated in tumor invasion and metastasis. Moreover, vitamin D supplementation has been shown to produce increased levels of cellular antioxidant enzymes such as thioredoxin rereductase 1, superoxide dismutase (SOD) 1 and 2, and glucose 6 phosphate dehydrogenase necessary for the maintenance of reduced glutathione, as well as other antioxidant enzyme pathways. Additional research studies have suggested a chemoprotective effect of high-level SOD enzymes [Oberley, 2001, 2005; St Clair et al. 2005]. Additional research suggests that upregulation of Mn++SOD may be an important target for anticancer therapeutics [Duan et al. 2003; Pani et al. 2004; Venkataraman et al. 2005]. For example, overexpression of Mn++SOD caused the suppression of glioma and SV40 transformed lung fibroblasts [Zhong et al. 1997; Oberley, 2001, 2005]. In conjunction with promising clinical data on the chemopreventive effects of agents with anti-inflammatory/antioxidant properties, these data support the use of this therapeutic approach in long-term maintenance therapy.
Not all research findings support a protective anticancer effect of vitamin D supplementation. The North Central Cancer Treatment Group randomized controlled trial (RCT) data reported by Pritchard and colleagues concluded that continuous baseline levels of 25-hydroxyvitamin D less than 72 versus greater than 72 nmol/liter did not affect event-free survival outcomes in patients with breast cancer [Pritchard et al. 2008].
RCT data from The Women’s Health Initiative reported by Chlebowski and colleagues showed no reduction in breast cancer risk in 36,282 postmenopausal women who received 1000 mg of calcium and 400 IU of vitamin D for a mean duration of 7 years compared with the placebo group [Chlebowski et al. 2012]. In addition, the study also showed no correlation between vitamin D intake, baseline 25-hydroxyvitamin D levels and breast cancer risk. Since the study did not evaluate the effects of vitamin D and calcium supplementation separately, one cannot know whether vitamin D intake only might have produced a different result. In addition, the protective effect of vitamin D may require serum baseline levels of at least 75 nmol/liter, which would require approximately 1700–2000 units of daily consumption, almost double that assessed in this RCT. Moreover, the study authors suggested that genetically determined high-level serum vitamin D levels may correlate with lower breast cancer risk.
Results of the Life After Cancer Epidemiology (LACE) cohort study of the effects of vitamin C, vitamin E and combination carotenoid supplementation on breast cancer mortality showed that vitamin C and E intake before and after breast cancer diagnosis was associated with 20% decreased recurrence risk, 32% decreased breast cancer mortality and 22% reduced risk of all-cause mortality [Greenlee et al. 2012]. The study authors speculated that this therapeutic benefit may result from their effects on oxidative stress pathways. The results showed that vitamin C or vitamin E supplementation reduced the risk of disease recurrence. Interestingly, the data also showed that vitamin E supplementation correlated with a better protective effect against disease recurrence in patients who also received radiation therapy and hormone therapy; moreover, the use of antioxidant supplements did not adversely affect outcome regardless of treatment modality, a conclusion that contradicts other research studies, suggesting that antioxidant supplementation may actually subvert the clinical benefits of chemotherapy and radiation therapy (see below). In contrast, the use of combination carotenoids was associated with increased recurrence risk and twofold increased mortality, a finding supporting other clinical trial data that indicated that carotenoid use may be contraindicated in cancer prevention. Moreover, the use of combination carotenoids was associated with higher mortality risk in patients who received chemotherapy, radiation or hormone therapy as opposed to patients who did not consume these supplements as part of these therapy regimens. The study authors concluded that consumption of vitamins C and E by women diagnosed with breast cancer was associated with better prognosis, measured by lower mortality and recurrence rates. Likewise, the results of the Shanghai Breast Cancer Survival Study indicated that consumption of vitamins E, C or multivitamins within 6 months of breast cancer diagnosis correlated with 22% decreased recurrence rate and 18% decreased mortality [Nechuta et al. 2011].
Despite accumulating evidence to suggest an important role of antioxidants in cancer prevention, there has been much controversy over their potential therapeutic applications based on the fact that these agents block free radical formation as an important mediator of their anticancer effects [Watson, 2013]. Although free radicals may function as important intracellular carcinogens at high levels, the formation of free radicals is required to elicit the cytotoxic tumor cell killing activities of many standard chemotherapy drugs as well as radiation [Block, 2004; D’Andrea, 2005]. Among the barriers to conventional therapy presented by the tumor microenvironment, it is well known that hypoxia inhibits effective radiation killing, due to its limiting effects on the production of ROS [Komarova and Wodarz, 2005]. In addition, chemotherapy resistance associated with low oxygen concentrations affects the activity of drugs such as melphalen, bleomycin and etoposide, all of which require molecular oxygen for their cell killing effects. Research on chemotherapy resistance further suggests that stem cells with low concentrations of ROS may be an important cause of treatment failure [Achuthan et al. 2011; Koukourakis et al. 2006]. Hypoxia can also induce cell cycle arrest and resistance to apoptosis, both of which can dramatically decrease the efficacy of drugs that target proliferating cells [Michor et al. 2005; Kozusko and Bourdeau, 2007]. That said, the long-term preventive and therapeutic approach presented in this paper does not suggest the use of antioxidants as adjuvants to acute cytotoxic chemotherapy or radiation, but rather their potential impact as a maintenance therapy in individuals at high risk for developing cancer and in patients diagnosed with cancer to prevent disease progression and recurrence.
Conclusion
Taken together, on a mechanistic level, clinical data suggest that some of the apparent chemopreventive properties of anti-inflammatory and antioxidant agents may instead act on pre-established incipient malignancies in which these agents block tumor progression and the onset of overt disease. The paradigm that prevention and treatment are interconnected therapeutic approaches is based on the apparent relationship between physiological conditions associated with the development of incipient cancer, progression to systemic disease and disease recurrence. The conception of long-term maintenance therapy approaches to cancer treatment involves the hypothesis that their cancer preventive and therapeutic effects may involve similar mechanisms. In other words, based on the redundancy of biological processes involved in cancer development, spread and recurrence, cancer prevention versus treatment may not be entirely appropriate or necessary distinctions; rather, they may represent approaches that target disease progression along a continuum, from early stage preneoplastic lesions to invasive cancers capable of metastatic spread, to re-emergence of residual disease. By altering the tumor-promoting environment, either in premalignant, active or postmalignant conditions, these so-called preventive agents may actually have a therapeutic function to maintain a systemic equilibrium that is resistant to tumor development, metastasis and disease recurrence. For example, recent studies by Walsh and colleagues on tumor cell metabolism using optical metabolic imaging showed that treatment of HER2/neu-positive human breast cancer cell lines and human/mouse xenografts with the monoclonal antibody trastuzumab decreased the redox ratio of NADH/FAD in tumor cells that responded favorably to the drug, but not in treatment-resistant cells [Walsh et al. 2013]. This finding implicates effects on redox signaling in the activity of this gene-targeted drug.
The successful clinical application of traditional chemotherapy protocols versus the more recently developed gene-targeted approaches and the long-term maintenance therapy approaches presented here may depend on appropriate designations of human cancer types most likely to benefit from some or all of these therapeutic approaches based on genetic, tissue and behavioral distinctions that may affect clinical outcome in response to specific therapeutic modalities. Critical etiological factors that may define distinctive clinical response parameters may include genetic differences between sporadic versus inherited cancers; tumor initiation factors associated with the development of hematological cancers versus solid tumor malignancies; and genetic and tissue origins of pediatric cancers versus common adult onset carcinomas.
For example, cancers resulting from inherited genetic predispositions, such as BRCA-1 breast cancer, apc-associated familial colon cancer and others may be more amenable to specific gene targeted approaches than noninherited ‘sporadic’ cancers that originate from multiple and diverse genetic lesions, a category that includes many adult-onset cancers. Likewise, leukemias/lymphomas resulting from specific translocations, inversions or other genetic lesions that are an aberrant byproduct of hematopoietic differentiation may profit from genetic screening and targeted therapeutic applications, as illustrated by the success of Gleevec for CML and rituxumab (Rituxan Genentech/Roche), a monoclonal antibody that specifically targets CD20-positive leukemias and lymphomas. Yet another etiological category encompasses childhood malignancies resulting from dysregulated embryonic genes, resulting in abnormal proliferation of embryonically derived cells that may be amenable to cytotoxic phase-specific and no-specific chemotherapy as well as gene-targeted approaches that counteract the proliferative effects of dysregulated embryonic genes. In contrast, many common adult carcinomas may develop more slowly over many years, emerging from a dysregulated genetic background that ultimately alters local and systemic parameters responsible for malignant progression. Cancers of this type may require multifaceted systemic therapeutic approaches that target both acute parameters of abnormal cell proliferation with cytotoxic chemotherapy and long-term chronic broad-spectrum parameters of disease progression.
Though we may not fully understand the complex processes that underscore the biological evolution of many complex adult-onset malignancies, this therapeutic paradigm expands the goals of traditional acute cancer therapy approaches to long-term clinical approaches designed to prevent or even reverse the systemic outcome of progressive disease.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.