
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease, characterized by progressive worsening of motor symptoms. The primary pathological hallmark of PD is the degeneration of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies, which are primarily composed of α-synuclein (α-syn) aggregates. Both α-syn and various neurotransmitters, including catecholamines (catechols), play crucial roles in the pathogenesis of PD, although the precise pathogenic mechanisms remain incompletely understood. The crosstalk between neurotransmitters and α-syn is intricate and multifaceted. Pathological α-syn disrupted neurotransmitters’ homeostasis by impairing release and reuptake of neurotransmitters, with specific modulation of catecholaminergic and glutamatergic systems. Conversely, neurotransmitters, especially catechols, covalently modify α-syn. Such modifications significantly influence α-syn aggregation dynamics and alter its neurotoxic properties. However, determining whether these interactions induce synergistic toxicity or confer neuroprotection remains controversial. Emerging evidence suggests other neurotransmitters like serotonin and γ-aminobutyric acid may also modulate α-syn aggregation and PD progression, though their roles require further investigation. Understanding these interactions is crucial for developing novel diagnostic and multi-target therapeutic strategies.
Plain language summary
Parkinson’s disease (PD), a brain disorder that causes worsening movement problems, involves two critical factors: a protein called α-synuclein and brain signaling molecules called neurotransmitters. While scientists know both play a role in the disease, exactly how they influence each other remains unclear. This research explores their complex relationship and its implications for improving patient care. In PD patients’ brain, two key changes: the buildup of harmful aggregates of α-synuclein (called Lewy bodies) inside brain cells, and disruptions in neurotransmitters, which are essential for smooth movement and other symptoms. The review reveals that these issues are deeply connected. For example, abnormal α-synuclein interferes with how neurotransmitters are released and recycled, creating communication breakdowns between nerve cells. At the same time, neurotransmitters like dopamine can chemically modify α-synuclein, either speeding up or slowing down its ability to form toxic aggregates. Why this matters? Understanding these interactions could lead to earlier diagnosis by identifying specific patterns in how α-synuclein and neurotransmitters behave. It could also improve treatments. Current therapies focus on replacing lost dopamine, but strategies targeting both α-synuclein aggregates and neurotransmitter imbalances might work better. For example, drugs that block harmful α-synuclein modifications while restoring healthy neurotransmitter levels could slow disease progression.

Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer’s disease and statistics predicted that by 2040, the number of PD cases among adults over 65 years old would exceed 12 million.1,2 PD patients often present as progressive movement disorders characterized by tremor at rest, rigidity, akinesia (or bradykinesia), and postural instability as well as a series of heterogeneous non-motor symptoms. 3 The clinical diagnosis of PD is mainly based on motor symptoms and other supporting evidence. However, the gold standard for diagnosis is the pathologic features of postmortem brain tissue sections, manifested by the loss of dopaminergic neurons in the substantia nigra (SN) and the presence of neuronal inclusions primarily composed of α-synuclein (α-syn) aggregates, known as Lewy bodies.4,5 Consequently, disturbances in α-syn and neurotransmitters, led by catecholamines (catechols), have long been recognized as vital factors in the pathogenesis of PD, although the pathogenic mechanisms are complex and still not entirely understood.
α-syn, encoded by the SNCA gene of chromosome 4, is a cytoplasmic protein composed of 140 amino acids, primarily located at the presynaptic terminals of neurons. 6 It is divided into three regions based on its structural features, with each region playing a unique functional role: the N-terminal region (residues 1–60), containing seven recurring imperfect repeats (KTKEGV), is positively charged and highly susceptible to gene mutation and responsible for membrane binding with α-helical structure; the non-amyloid-β component region (NAC, residues 61–95), a key component involved in aggregation process via β-sheet structure; and the C-terminal region (residues 96–140), negatively charged and prone to post-translational modifications (PTM).7,8 The main function of α-syn is associated with synaptic vesicle trafficking and thus has an impact on neurotransmitters’ recycle process. 9 Under physiological conditions, α-syn exists as disordered protein monomers 10 or aggregation-resistant helical tetramers 11 and transforms into α-helices when bound to lipids. In the PD’s pathogenesis, α-syn may experience misfolding and subsequently pathological aggregation: misfolded α-syn forms soluble oligomers with β-sheet structure that progressively polymerize into insoluble fibrils and propagate in a prion-like manner. The insoluble fibrils eventually form Lewy bodies with other substances deposited within neurons. Moreover, the aggregation process can be further divided into three phases based on the aggregation kinetics and appears as an S-shaped curve: a lag phase, a growth phase, and a plateau phase, involving various intra- and inter-interactions in three regions. 12 Among different structural forms of α-syn, oligomers are widely recognized as the main toxic form of PD pathogenesis.13,14 Mechanisms of its toxicity may be summarized as loss of normal function, destruction of cellular structure or function, and acquisition of other toxicity.15–17 Additionally, there are many factors involved in the modulation of α-syn toxicity, and investigating various influences on α-syn aggregation process is vital to understand its role in the pathogenesis of PD.
In addition to Lewy bodies formed by α-syn aggregates in the brains of PD patients, depletion of dopaminergic neurons, resulting in abnormal changes in neurotransmitters’ level, mainly catechols, has become another vital feature of PD and the key pathogenic mechanisms. Catechols are mainly consist of dopamine (DA) and its derivatives and disorders in catechols’ metabolism results in imbalance of catechols’ homeostasis in PD. 18 DA, an important precursor of other catechols, is synthesized through hydroxylation of L-tyrosine by tyrosine hydroxylase (TH), followed by decarboxylation by aromatic acid decarboxylase (AADC). Synthesized DA will be transported into vesicles by vesicular monoamine transporter 2 (VMAT2) for reserve.19,20 Nonetheless, cytoplasmic DA is susceptible to catabolism via a series of oxidative processes, including auto-oxidation and enzyme-catalyzed oxidation, yielding various DA oxidation products and byproducts, some of which, particularly 3,4-dihydroxyphenylacetaldehyde (DOPAL), proposed to be neurotoxic.15,21,22 Stimulated by motor or other stimulus signals from the brain, restored DA will be released into the synaptic cleft by exocytosis to function. At the end of its activity, released DA will be reuptake into the cytosol via transporters to maintain homeostasis. Disorders in DA metabolism, including biosynthesis, vesicle trafficking, and catabolism, triggers intracellular and extracellular catechols’ imbalance which has been recognized as a major reason for the motor dysfunction of PD patients. In addition to abnormalities in catechols, research showed that abnormalities in other neurotransmitters, such as glutamate, γ-aminobutyric acid (GABA), and serotonin or 5-hydroxytryptamine (5-HT) could result in heterogeneous non-motor symptoms in the early and late phases of PD.23,24 Thus, focusing on aberrant alterations in different neurotransmitters and their influencing factors is helpful to gain a comprehensive understanding of the mechanisms underlying the heterogeneous clinical symptoms of PD and the progression of the disease.
α-syn pathological aggregation and disturbances of neurotransmitters, primarily catechols, are not two independent pathways in the pathogenesis of PD. Rather, these two pathogenic pathways impact with each other in multiple ways, leading to the onset and progression of PD. Emerging evidence reveals that these processes interact in a bidirectional manner through multiple molecular mechanisms, collectively driving disease occurrence and progression. This review systematically examines the reciprocal pathological relationships between α-syn aggregation and neurotransmitters’ homeostasis, with particular emphasis on evaluating whether these interactions predominantly exert neurotoxic versus neuroprotective effects. Furthermore, we demonstrated current clinical translation advances that leverage this pathophysiological crosstalk to develop novel diagnostic and therapeutic strategies for PD management. It is hoped that it may assist in advancing the understanding of PD pathogenesis and facilitating the development of innovative diagnostic strategies and targeted therapeutic interventions.
α-syn-neurotransmitter crosstalk in PD’s pathogenesis
α-syn-mediated regulation of neurotransmitter homeostasis
The presynaptic localization of α-syn suggests that this protein serves as a critical modulator of neurotransmitter signaling dynamics. Accumulating evidence supports its dual physiological-pathological role in maintaining synaptic vesicle trafficking integrity while contributing to neurotransmitter dysregulation in PD’s pathogenesis. Mechanistic studies revealed that α-syn abnormalities disrupted synaptic vesicle recycling (neurotransmitter release and reuptake processes) through multiple mechanisms, as schematized in Figure 1.

The general effects of α-syn on neurotransmitters. The general effects of α-syn on neurotransmitters trafficking including: (a) the negative modulation role in expression and activity of VMAT2, (b) interfere in SNARE complex formation or induction of severe vesicles clustering, (c) block the vesicles in the docking area, (d) inhibition of the amount of neurotransmitters release, and (e) induction of MET shuttle from membrane surface to cytoplasm.
α-syn-induced dysregulation of neurotransmitter release
Neurotransmitter exocytosis proceeds through four sequential processes, including clustering of neurotransmitter-containing vesicles from the reserve pool to the readily releasable pool, vesicle docking, calcium-mediated membrane fusion, pore opening, and exocytosis. 25 Pathological α-syn accumulation disrupts this cascade through the following mechanisms:
Emerging evidence indicated that α-syn overexpressed caused ultrastructural abnormalities of synapses. Transgenic mouse models overexpressing α-syn exhibited reduced synaptic vesicle density accompanied by vesicular size heterogeneity and decreased presynaptic protein expression, 26 which may be the structural basis for disorders in synaptic vesicle trafficking.
Mechanistically, α-syn exerts multiple regulatory effects throughout the synaptic vesicle cycle. Prior to the initiation of the vesicle trafficking process, neurotransmitters are transferred into vesicles by the specific transporters for storage and remain in the reserve pool, which is significantly affected by the expression and activity of vesicles’ membrane transporters. The expression and activity of vesicular neurotransmitter transporters are critically modulated by α-syn. Particularly, the uptake activity of VMAT2, responsible for DA, norepinephrine (NE), and serotonin (5-HT) packaging, is significantly inhibited by both wild-type (WT) and A53T mutant α-syn.27–29 More specifically, α-syn aggregates down-regulated VMAT2 expression, while α-syn monomers did not exhibit such inhibitory effect.30,31 Subsequently, neurotransmitter-containing vesicles move from the storage zone (reserve pool) to the active zone (readily releasable pool). Then, vesicles dock in the active zone, awaiting initiation of membrane fusion and the release of neurotransmitters via exocytosis. In vitro studies demonstrated that both WT and A30P mutant α-syn impaired late-stage vesicle trafficking through (1) physical obstruction of vesicle docking sites, and (2) suppression of quantal neurotransmitter release. Importantly, these modulatory effects exhibited cell-type specificity. 32
Exocytosis involves the coordinated activity of multiple protein complexes, with the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex serving as the central molecular machinery mediating synaptic vesicle fusion. α-Syn inhibited SNARE-mediated vesicle fusion in a concentration-dependent manner by dual modulation of complex expression and functionality. Initially, α-syn was found to co-localize with some of the SNARE complex proteins and could downregulate the expression of some SNARE complex components.26,33–35 Under physical conditions, α-syn oligomers maintain dynamic interactions with synaptobrevin-2 (VAMP2), either through direct binding or via cooperative lipid-associated monomer interactions that facilitate SNARE complex assembly. Nonetheless, excessive α-syn contributed to covering the binding site of VAMP2 for SNARE complex formation and causing severe vesicle clustering, which in turn decreased the number of vesicles in the release pool and eventually decreased the neurotransmitters’ release. Notably, mutation-specific variations in clustering efficacy have been reported, with the E46K variant exhibiting the most pronounced clustering effect and A30P showing minimal impact. Further, E46K oligomers displayed unique membrane-interaction properties, significantly reducing bilayer mobility and increasing permeability. However, it is still controversial whether α-syn oligomers or monomers are more effective at disrupting the SNARE complex’s ability to facilitate vesicle fusion.36–39 In addition, researchers proposed that there was a recycling pool shuttling between the reserve pool and the readily releasable pool, which contained about 10% of the total number of vesicles. It was shown that α-syn could also inhibit neurotransmitters’ release by diminishing the size of the recycling pool.34,40 Collectively, these findings establish α-syn as a multifaceted negative regulator of synaptic transmission, providing a comprehensive framework for understanding the pathophysiological role of α-syn in synaptic dysfunction.
α-syn-induced dysregulation of neurotransmitter uptake
Neurotransmitter reuptake represents a critical regulatory mechanism for synaptic transmission termination and presynaptic vesicle replenishment, which depends strongly on the level of specific transporters on the synaptic surface via the affinity and the uptake rate of transporters. The most prominent effect of α-syn on transporters was a highly homologous monoamine transporter family (MET), including dopamine transporters (DAT), norepinephrine transporters (NET), and serotonin transporters (SERT). Despite the highly similar structure within the MET family, the effects and mechanisms of α-syn on these transporters vary.
For DAT, significant co-localization of DAT with α-syn underlies their interaction. Structural analyses revealed that the NAC domain of α-syn interacted with the C-terminal region of DAT, facilitating its internalization from the plasma membrane to cytoplasmic compartments. α-Syn aggregates displayed similar effects by retaining DAT in inclusions. Moreover, the activity of DAT was also attenuated by α-syn by diminishing uptake velocity, with the strongest inhibition effect in A30P α-syn, while a negligible effect in A53T α-syn. The disparity in inhibitory effects by different α-syn genotypes may correlate with the variation in their binding affinity with DAT.41–45 Microtubule disruption experiments further suggested cytoskeletal involvement in α-syn-DAT interactions. 46 Still, conflicting evidence suggested that α-syn enhanced the DAT number in the plasma membrane, increasing reuptake of DA. 43 The role of α-syn in modulating DAT remains debated. 47
For NET and SERT, α-syn was able to bind and act as a negative modulator. Evidence showed that overexpressed α-syn decreased the density of NET and SERT at the membrane surface, decreasing neurotransmitter uptake capacity without altering substrate binding affinity.27,48–50 Notably, the structural homology among MAT (NET, SERT, and DAT) implied cross-regulatory effects, where pharmacological inhibition of one transporter may compromise functional compensation by others. 47
Moreover, α-syn also modulated vesicle recycling by inhibiting phospholipase D activity, an important enzyme involved in vesicle formation and budding.51–53 Overall, the modulation of α-syn on the synaptic vesicle trafficking process is complex and diverse, abnormal α-syn in PD upsets the equilibrium of intracellular and extracellular neurotransmitters. Despite that α-syn manifests pathological inhibition of release and reuptake process in general, differential expression of α-syn in different neurons and other co-factors lead to that not all neurotransmitters display a decrease trend in the PD,54,55 suggesting that the recycle of synaptic terminal neurotransmitters undergoes complex regulation, with the negative regulation by α-syn being only one important mechanism, there may be other mechanisms that outweigh α-syn in facilitating the recycle of such neurotransmitters.
Neurotransmitter-specific modulation by α-syn
Beyond the broad influence of α-syn on neurotransmitters’ release and reuptake mechanisms, accumulating evidence reveals that pathological α-syn accumulation exerts specific disruptive effects on certain neurotransmitter systems.
The specific effect of α-syn on the catecholaminergic system
One of the most striking features of PD is depletion of dopaminergic neurons and a decrease in DA levels, thus the vast majority of studies on the impact of α-syn on neurotransmitters focused on catechols. While the general effects of α-syn on vesicular trafficking are well-documented, recent investigations uncover its specific interference with catechols’ biosynthesis pathways.
As an important precursor substance for other catechols, the synthesis of DA is important for maintaining other catechols’ homeostasis. DA is converted from tyrosine with the help of two important enzymes, TH and AADC, with TH serving as the rate-limiting enzyme in DA synthesis. 56 TH was negatively regulated by α-syn.
First, experimental models revealed wild-type α-syn overexpression suppressed TH expression by interfering with transcription factor, though the inhibitory potency varied significantly among α-syn mutants.33,57,58 Additionally, α-syn affects the activity of TH. 56 A study showed that α-syn was structurally similar to 14-3-3 protein, which was associated with TH activation. Nonetheless, in contrast to 14-3-3 protein, α-syn inhibited the activation of TH. Moreover, researchers proposed that TH activity was affected by phosphorylation. Studies showed that overexpressed wild-type or mutant α-syn inhibited TH activity via decreasing its phosphorylation either through direct interaction or indirect interaction with ERK, an enzyme required for TH phosphorylation.59,60 Intriguingly, this inhibitory effect appears attenuated during α-syn aggregation processes.60,61 Moreover, another study found phosphorylated Ser129 α-syn variants conversely exhibited TH-activating properties in vitro, and such phosphorylation was highly prevalent in PD.62,63
The regulatory complexity extends to NE biosynthesis, where α-syn interfered with dopamine β-hydroxylase transcription, potentially disrupting DA-to-NE conversion. 64 As such, these findings suggested that α-syn was a multifaceted modulator of catechols’ biosynthesis, with effects contingent upon its conformational state, PTMs, and intracellular concentration. The regulatory mechanisms underscore the importance of α-syn species analysis when evaluating its pathophysiological contributions to neurotransmitter disorders.
The specific effect of α-syn on glutamatergic system
Apart from depletion of dopaminergic neurons, glutamatergic pathway dysregulation was also identified as a critical contributor to the motor dysfunction and other symptoms of PD. The vesicular glutamate transporter (VGLUT) family, consisting of VGLUT1, VGLUT2, and VGLUT3, is responsible for packaging glutamate into vesicles. VGLUT1 and VGLUT2 display complementary distribution within the brain and also serve as specific markers for glutamatergic neurons.65,66 Postmortem analyses revealed regional VGLUT1/VGLUT2 expression abnormalities in PD brains, suggesting their involvement in disease progression. 65 Further, though the widespread distribution of α-syn in glutamatergic neurons, it demonstrated subtype-specific modulatory effects on VGLUT1 and VGLUT2 systems.
For VGLUT1, α-syn preferentially localizes to VGLUT1 axon terminals in cerebral cortical neurons. α-syn fibrils caused abnormal changes in synaptic terminal morphology and increased vesicle clustering. Additionally, the presence of α-syn aggregates disrupted VGLUT1 cortico-basolateral amygdala synaptic glutamatergic transmission, resulting from a reduction in the number of readily releasable synaptic vesicles or synaptic vesicle release sites.67,68 Strikingly, α-syn exhibits paradoxical effects across VGLUT subtypes. While destabilizing VGLUT1 synapses, it upregulated VGLUT2 expression in striatal neurons following α-syn preformed fibril administration. The upregulation of expression exerted a neuroprotective effect, as the increased influx of glutamate into the vesicle amplified the pH gradient of the vesicle, facilitating the sequester of DA into the vesicles and reducing reactive oxygen species (ROS) caused by cytoplasmic accumulated DA.69–71
In other non-neuronal brain cells, such as astrocytes, α-syn exerted a distinct detrimental effect. Oligomeric α-syn increased vesicular glutamate release in a Ca2+-dependent way. Moreover, α-syn oligomers could also activate glutamate receptor (NMDA receptor), indirectly and directly, to cause synaptic spine loss. 72 These findings underscore the cell type-specific complexity of α-syn-mediated glutamatergic modulation.
Neurotransmitter-mediated regulation of α-syn dynamics
The α-syn-neurotransmitter interaction constitutes a reciprocal pathogenic loop in PD. While α-syn profoundly impacts the homeostasis of several neurotransmitters, reports revealed neurotransmitters, particularly catechols, conversely modulated α-syn pathology through distinct molecular mechanisms.
The effect of catecholamines on α-syn—α-syn catecholamization
Given that the selective loss of catecholaminergic neurons in PD, predominantly dopaminergic neurons, along with disorders in catechols’ metabolism, studies of neurotransmitters’ effect on α-syn have primarily focused on catechols. Catechols mainly regulate the α-syn aggregation process, along with other effects.
The effect on α-syn aggregation—promotion of oligomerization and inhibition of fibrillation
In the early 21st century, evidence elucidated that of the 15 substances screened for inhibitory effects on α-syn fibrillation, all but 1 were catechols, 73 characterized by the prolonged lag phase74,75 or the growth phase 76 during the α-syn aggregation process. The dual roles of catechols in the α-syn aggregation process were the promotion of oligomerization and the inhibition of fibrillation, which were often affected in a time- and concentration-dependent manner.74–82 Further, the mechanisms and interaction sites underlying the inhibitor effects were complicated and varied among studies, including (Figure 2 and Supplemental Table 1):
(1) Oxidation of α-syn methionine (Met) to Met-sulfoxide. The formed Met-sulfoxide increased negative charges at C-terminus to strengthen the intra-molecular hydrophobic interactions and long-range electrostatic interactions of α-syn, inducing a greater solvent exposure of the NAC domain.74,80,82–93
(2) Covalent interaction with α-syn lysine (Lys) (mainly in the N-terminal and NAC region). α-syn-Lys-catechol adducts reduced the positive charges at N-terminal and increased the intra-molecular hydrophobic interactions.74,75,77,78,80,82,83,87,92–96
(3) Non-covalent hydrophobic interaction with Y124EMPS128 motif in the C terminus of α-syn.75,97–101
(4) The electrostatic interactions with glutamate 83 (E83) in the NAC region, decreasing surface-exposed hydrophobic clusters.75,97
(5) Inhibition of fibril ends growth. 76

Possible mechanisms of interactions between α-syn and catechol(s). Upper lane: the structure and interaction sites between α-syn and catechol(s). Lower lane: amino acid sequence of α-syn and possible mechanisms of interactions between α-syn and catechol(s). (a) Oxidation of Methionine to Met-sulfoxide. (b) Covalent interactions between α-syn Lys and catechol(s) via Schiff base reaction. (c) Non-covalent interaction between Y125EMPS129 motif of α-syn and catechol(s).
Herein, the effects of catechols on α-syn aggregation are extremely intricate, and it is possible that three regions of full-length α-syn are involved in such interactions with different mechanisms. More importantly, the oligomers induced by catechols could not polymerize into fibrils like α-syn self-aggregation (“on-way aggregation”), called “off-way aggregation,” reflecting the inhibition effect on α-syn fibrillization indirectly (Figure 3). Additionally, some studies proposed that during the off-way aggregation process, large oligomers and small oligomers (dimers or trimers) emerged simultaneously through kinetically distinct processes.83,99 Moreover, α-syn-catechol modification exhibited enhanced oligomer stability,96,98,102 as well as fibrils disaggregation capabilities.103,104 However, in the presence of lipid structure, the pro-oligomerization capabilities underwent significant attenuation, suggesting environmental modulation of α-syn-catechol interactions.74,89,105
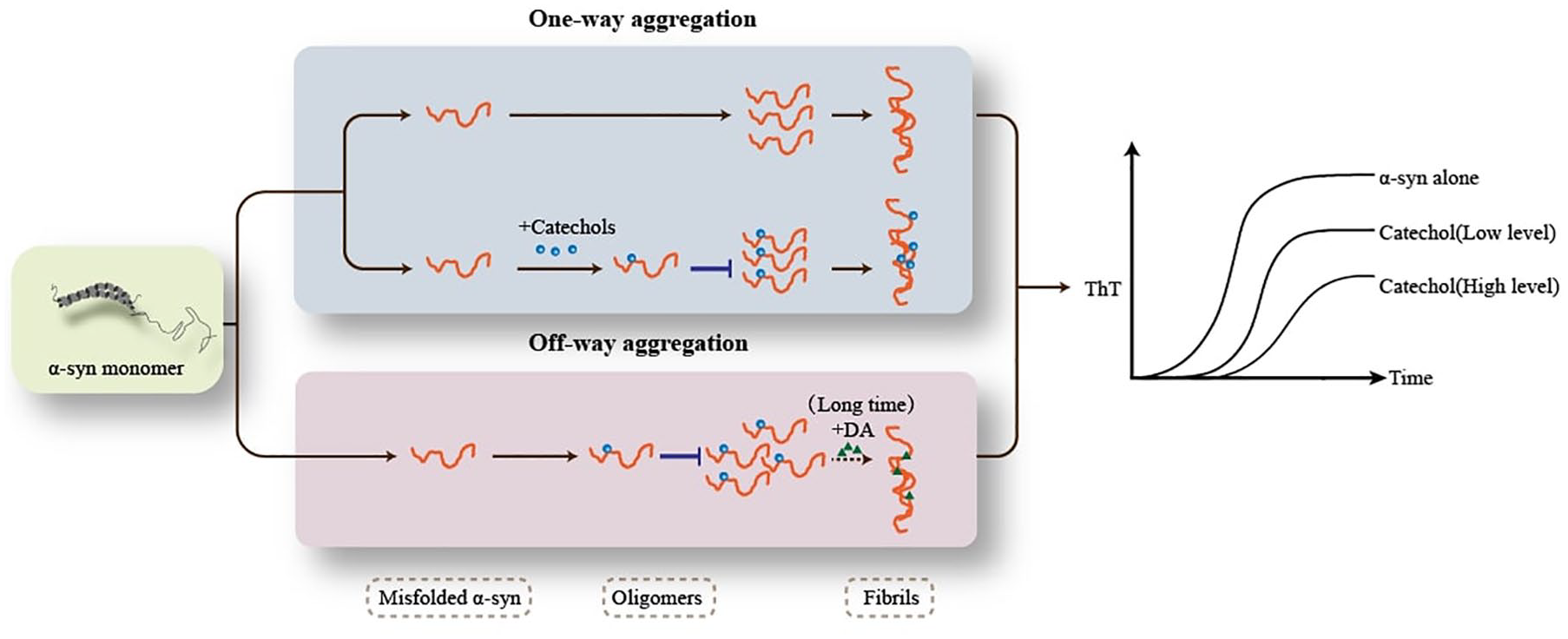
The on-way aggregation and off-way aggregation of α-syn. Blue lane: On-way aggregation process, including α-syn self-aggregation (upper) and α-syn-catechol aggregation (lower), is from misfolded α-syn monomers to β-sheet and ThT-positive fibrils, with the oligomers as intermediate products. Pink lane: Off-way aggregation process, or α-syn-catechol aggregation, is from misfolded α-syn monomers to ThT-negative oligomers (end products). Only in the presence of DA, α-syn monomers aggregated into fibrils after a long time. Right lane: In the α-syn alone, ThT experiment shows the strongest fluorescence. In the presence of catechols, ThT fluorescence decreases with increased catechols’ level.
Notably, the inhibitor effects vary markedly among catechols’ species.75,76,79,81,104,106 For example, despite most research showing the significant inhibitor effect of DA, the controversy over whether DA has the ability to regulate α-syn aggregation persists. Several investigations proposed that the role of DA in promoting oligomerization was negligible or incapable of inducing α-syn aggregation, even under in vitro conditions well above physiological DA concentrations (up to 1400 μM).15,77,79,81,88 And further evidence proposed that the role in inducing oligomerization of DA could be suppressed by antioxidants or reducing agents, combined with the negligible amount of α-syn-DA adducts and the susceptibility of DA to enzyme-catalyzed oxidation or autoxidation, it is suggested that DA oxidation products, rather than DA itself, were involved in the α-syn aggregation.73,87,88,96,99
DA oxidation, or catabolism, occurs via two pathways: enzyme-catalyzed oxidation and self-oxidation. During enzyme-catalyzed oxidation, DA is firstly degraded by mitochondrial monoamine oxidase (MAO) into DOPAL, a neurotoxic metabolic intermediate, which is subsequently converted to the non-toxic 3,4-dihydroxyphenylacetic acid (DOPAC) by aldehyde dehydrogenase (ALDH). 19 DA stored in vesicles is stable but cytoplasmic DA is highly susceptible to degradation. In PD, vesicular leakage and impaired ALDH activity lead to pathological accumulation of cytoplasmic DOPAL, a potent driver of α-syn oligomerization.107,108 Previous research found that catechols, including DA oxidants, could prevent α-syn aggregation into fibrils, while increasing the formation of oligomers. 99 Among the DA oxidation products, studies revealed that DOPAL exerts the strongest effect in promoting α-syn oligomerization (functioning at concentrations as low as 1.5 μM), 79 with higher-order aggregates emerging in a dose-dependent manner.74,77–79 Studies emphasized the significant toxic effects of DOPAL, including its influence on the α-syn aggregation process and proposed “the Catecholaldehyde Hypothesis.”21,22 Similar to DA, the pro-oligomerization ability of DOPAL could also be suppressed by antioxidants. 109
Additionally, evidence suggested that, in addition to DOPAL, other DA metabolites, such as DOPAC, the non-toxic DA metabolic end-product, could also exert inhibitory effects on α-syn aggregation. The mechanisms of DOPAC on α-syn aggregation also depended on its concentration. Research showed that a low level of DOPAC (DOPAC:α-syn ⩽ 1:1) influenced α-syn fibrillization indirectly through non-covalent binding of DOPAC-quinone (DOPAC-Q) to α-syn. However, when DOPAC:α-syn ratio was greater than 3:1, DOPAC could affect aggregation directly, as well as DOPAC-Q covalent modification and α-syn Met oxidation. 80 Besides the main DA metabolites mentioned above, other catechols, such as levodopa (L-DOPA), dopaminochrome, epinephrine (E), and NE also exhibit similar effects on α-syn fibrillization.75,88,97,99 Different from catechol-induced non-amyloidogenic oligomers, oligomers induced by NE displayed the amyloidogenic forms, which may be attributed to the different binding site on α-syn. 110
Further, α-syn aggregation modified by catechols (off-way aggregation) exhibits distinct properties compared to α-syn self-aggregation (on-way aggregation), as summarized in Table 1 and Figure 3. First, off-way aggregation does not produce fibrils (except for DA after prolonged incubation 101 ), while α-syn fibrils are the end products in the on-way aggregation process. Second, α-syn oligomers or fibrils formed by self-aggregation are often amyloidogenic with β-sheet structure, while in the presence of catechols, α-syn oligomers were characterized as non-amyloidogenic species, such as spherical or rod-shaped oligomers.38,75,89,99 Third, the majority of research proposed that α-syn oligomers triggered by catechols were often present with specific features, being ThT-negative, thermally stable, and sodium dodecyl sulfate (SDS)-resistant, different from ThT-positive and SDS-sensitive oligomers formed by α-syn self-aggregation or other pro-aggregation agents. 89 Fourth, α-syn oligomers were reported to increase membrane permeability by forming pores in the plasma membrane. Some studies revealed similar effects for α-syn-catechol oligomers,82,83 while other research suggested that α-syn-catechol oligomers may exhibit decreased or no neurotoxicity.38,76,86,111 More importantly, the species of oligomers induced by catechols also differed based on the types of catechols. Research showed that DOPAL was prone to form smaller oligomers (dimers or trimers), while DA usually promoted the formation of larger oligomers and even fibrils after prolonged incubation.74,112 In addition, larger α-syn-DOPAL oligomers appeared to be associated with increased neurotoxicity. 94 Lastly, DA and DOPAL showed different oligomerization capabilities, with DOPAL exhibiting a faster rate of inducing oligomerization. 112 Beyond the different inhibitory effects of catechols themselves, these effects varied across different α-syn mutants or PTM,15,77,94 and were amplified by the presence of divalent metal ions, such as Cu2+.77,81
Comparison between on-way aggregation and off-way aggregation.

DA, dopamine; DOPAL, 3,4-dihydroxyphenylacetaldehyde; SDS, sodium dodecyl sulfate.
The other effects of catechols on α-syn
In addition to the significant effects of catechols on inducing α-syn oligomerization, observations suggested that catechols exert other modulatory effects on α-syn.
First, a previous study showed that DA increased α-syn expression by de novo synthesis in SH-SY5Y cells. 113 Second, oligomers triggered by DOPAL enhanced cytotoxicity by impairing vesicles via a pore-forming mechanism. 83
Third, DOPAL not only induced α-syn oligomerization but also induced the quinonylation of α-syn through covalent binding to Lys on α-syn via DOPAL aldehyde group, a unique structure different from DA. Researchers speculated that quinonylation may be an intermediate step in the formation of oligomers. Nonetheless, the role of α-syn quinonylation induced by DOPAL in PD’s pathogenesis, as well as the exact relationship between α-syn quinonylation and oligomerization has not been elucidated yet.77,78
Fourth, studies showed that DA facilitated the release of α-syn oligomers into the extracellular space, which may increase cell-to-cell transmission of neurotoxicity.106,114 Lastly, one of the degradation pathways for WT α-syn monomers and dimers was through chaperone-mediated autophagy (CMA). Observations proved that high cytoplasmic DA level (>3 μM) in vitro exerted an inhibitory effect on CMA activity in the presence of α-syn, leading to an accumulation of α-syn-DA oligomers and blocking substrates translocation and degradation. 115
The other effects of neurotransmitters on α-syn: beyond catecholaminergic pathways
Despite catechols exerting a remarkable influence on α-syn, the impact of other neurotransmitters should not be overlooked. Current research reveals that serotonergic and GABA systems demonstrate distinct regulatory capacities on α-syn aggregation kinetics and secretory pathways.
Similar to the mechanism by which catechols inhibit α-syn aggregation, serotonin (or 5-HT) could bind to the C-terminal region of α-syn aggregates via electrostatic interactions, promoting the formation of intermediate species while interfering with fibril formation. 116 Additionally, 5-HIAL, the metabolite of 5-HT generated through MAO enzymatic activity, exhibited a similar capacity to promote α-syn oligomerization in vitro, comparable to that of DOPAL,48,117 indicating that monoamine aldehyde may be the key component promoting α-syn oligomerize.
Regarding GABA, a vital inhibitory neurotransmitter in motor signal transmission, though there was a lack of co-localized relationship between GABA and α-syn, researchers found that the secretion of α-syn was modulated indirectly by GABA. The study proposed that in cortical neurons, the secretion of α-syn was in an unconventional process, which is calcium-dependent and modulated by GABA. Activation of synaptic GABAB receptors by GABA release is required for modulating the secretion of α-syn, 118 which may help to better understand the abnormality in α-syn levels in the body fluid.
The interaction between catechols and α-syn: accomplices or helpers in PD?
The synergistic neurotoxic effects of catechols and α-syn
The remarkable mutual effects between α-syn and neurotransmitters, primarily catechols, play a significant role in the pathogenesis of PD. Studies demonstrated the interaction between catechols and α-syn may exert synergistic toxic effects, in other words, function as an accomplice in promoting PD pathogenesis.119–121
First and foremost, the neurotoxicity of catechols and α-syn was mutually dependent or reinforcing. On the one hand, DA can serve as a crucial source of endogenous ROS during metabolism, which can induce apoptosis. Some studies showed that cytoplasmic DA-induced toxicity depended on the presence of α-syn. α-Syn knock-out models displayed significant resistance to catechol-induced toxicity and neurodegeneration, suggesting that α-syn may be one of the key downstream substances in the DA pathogenesis.98,122–124 This dependency is further evidenced by α-syn’s ability to amplify DA-dependent ROS production, particularly through pathogenic mutations (A53T and A30P) that enhance pro-apoptotic effects.33,125 Studies suggested α-syn modulated VMAT2 expression and compromised DA sequestration, leading to increased cytoplasmic DA accumulation and subsequent oxidative stress.29,126
On the other hand, the neurotoxicity of α-syn displays catechol-dependent specificity. Pathological α-syn aggregates demonstrated selective toxicity in dopaminergic neurons while exhibiting neuroprotective properties in non-dopaminergic cells. This may be attributed to the vulnerable fibrils breaking into small toxic species in dopaminergic neurons, while stable non-toxic fibrils were retained in the non-dopaminergic neurons.102,125 Additionally, inhibition of endogenous DA expression may inhibit the formation of oligomers in vitro 38 and reduce α-syn-induced cytotoxicity.101,125,127 Further, catechols may act as an enhancer of α-syn toxicity. The most significant effect is the role of promoting α-syn oligomerization mentioned above, as oligomers are the main toxic form of α-syn. The formation of fibrils may represent a kind of neuroprotective reaction, indicating that catechols contribute to on α-syn’s pro-neurotoxic effect directly and indirectly.
More importantly, several positive feedback toxicity amplification mechanisms exist between catechols and α-syn. α-Syn elevated cytoplasmic catechols via disrupting DA storage, along with reduced detoxification by ALDH in PD. Increased catechols in turn triggered the formation of more α-syn-catechols modified oligomers, leading to further neurotoxicity.82,128 Notably, a redox-based amplification loop exists between DOPAL and α-syn. Research proposed that α-syn accelerates DOPAL oxidation, while the resultant oxidized DOPAL promotes methionine oxidation in α-syn, a critical PTM enhancing its aggregation propensity. 74
Last but not least, the interaction between α-syn and catechols also affected cell metabolism. Research showed that mutant α-syn (A53T, A30P) and DAT accelerated cellular energy depletion, which may be another important pathway in the PD pathogenesis.51,129
Further, other research suggested that there were indirect effects mediating synergistic neurotoxicity. First, in PD patients’ brains, it can be observed that highly activated asparagine endopeptidase (AEP), which was able to cleave human α-syn at N103. Truncated α-syn N103 fragment (α-syn N103) exhibited high-affinity binding to MAO-B, creating a pathogenic feedback loop where α-syn N103 enhanced MAO-B-mediated DOPAL production, which in turn activated more AEP, generating a toxic positive feedback loop.130,131 Additionally, according to the gut-first hypothesis in the early stages of PD, α-syn originates in the gut, with chronic inflammation caused by a disturbed gut microbiota involved in PD pathogenesis. Research found that the selective NE-depleting toxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) could induce PD by disrupting gut microbiota. Moreover, in SNCA mutant mice, the susceptibility to DSP-4 toxicity was increased and exacerbated DSP-4-induced gut inflammation, showing the synergistic pathogenic effects of NE and α-syn. 132
The neuroprotective effects of catechols and α-syn
Although the synergistic toxic effects of α-syn and catechols are well recognized, recent studies have proposed that this interaction also exerts neuroprotective effects. Researchers suggested that in the presence of catechols, the aggregation process was followed in a specific pathway, which differed from the α-syn pathological self-aggregation pathway (mentioned in section “The effect on α-syn aggregation—promotion of oligomerization and inhibition of fibrillation”).76,92,102 This alternative pathway was considered to possess non-toxic or even neuroprotective properties in the following ways.
To begin with, oligomers induced by catechols showed a decrease in cytotoxicity due to reduced disruption of plasma membrane integrity and function, as well as a reduction in ROS level, which may be due to fewer aggregates being on the membrane.38,75,76,86,92,133 Further, the interaction of DA and α-syn disrupted the seeding capacity of α-syn, suggesting that α-syn-DA modification confined the focal site and decreased the spread of disease, though seeding ability is neither necessary nor sufficient for toxicity. 98
In general, the synergistic toxic effects resulting from the interplay between α-syn and catechols were mainly through reciprocal promotion of oligomerization and ROS generation from catechol’s metabolism. In comparison with the neurotoxic interactions, the neuroprotective effects appear negligible. Nonetheless, most previous studies did not separate the catechol-induced oligomers from the oligomers generated by self-aggregation, it is therefore unreliable and adventurous to disregard the potential neuroprotective effects of catechols modification. Further, determining whether the relationship between a-syn and catecholamines acts as accomplices or helpers in PD pathogenesis remains challenging.
α-syn-neurotransmitter crosstalk: diagnostic and therapeutic implications
The dynamic interplay between α-syn and neurotransmitters, particularly catechol derivatives, has been extensively investigated over decades, yielding critical insights into PD’s pathogenesis while opening new frontiers in diagnostic and therapeutic innovation.
With the advancement of detect technique, nowadays an immunoprecipitation-mass spectrometry-based detection method enables the quantitative detection of various PTMs of α-syn, including dopamine binding, in plasma. The elevated concentration of dopaminergic α-syn in PD patients not only validated the modifying effect of DA on α-syn observed in previous in vitro experiments but also underscored the potential value of α-syn-dopaminylation detection in future diagnostic and prognostic tests. 134 Although the imbalance in neurotransmitters in PD is widely recognized, there have been few studies systematically integrate α-syn-neurotransmitter dynamics into clinically assessments.
Current therapeutic strategies increasingly emphasize multi-target approaches addressing PD’s multiple pathology, manifested through two aspects: (1) repurposing old drugs for new uses; (2) developing new drugs for modern needs.
First and foremost, neurotransmitter agents, especially “DA-supplement” therapy, as the first-line drugs for PD treatment, not only achieve symptomatic treatment by regulating imbalance in neurotransmitters but also begin to explore their effects on α-syn pathology in recent years. First-line dopaminergic agents, exemplified by L-DOPA combined with peripheral decarboxylase inhibitors like benserazide, now demonstrate dual therapeutic mechanisms: alleviating motor symptoms through basal ganglia dopamine replenishment while directly modulating α-syn pathology. A preclinical study showed that treating PD mice with L-DOPA/benserazide significantly inhibited the aggregation and even propagation of α-syn in brain. 135 Not only the direct effect of catechols on α-syn aggregation, researchers found SNCA hypomethylation occurs in the brain as well as in the peripheral blood of sporadic PD patients, while L-DOPA could increase SNCA DNA methylation in the blood in a dose-dependent manner. 136 Similarly, entacapone and tolcapone, two catechol O-methyltransferase inhibitors, and related catechol-containing compounds, were also found to influence fibrillization process by altering the structure of the aggregates and inducing into off-pathway non-toxic aggregation process. 137 Further, based on the structural features of catechols, researchers also engaged in the design and synthesis of novel therapeutic agents. A novel molecule, synthesized by DA and the naphthoquinone (NQ) molecule, could exert strong anti-aggregation and disruption fibrils effects through the synergistic effect of α-syn-DA binding and self-assembly inhibiting by NQ structure.138,139 Also, other DA-supplement agents, including MAO inhibitors140,141 and DA agonists 142 have demonstrated anti-α-syn fibrillization effects in preclinical investigations. Beyond dopaminergic systems, glutamatergic modulators such as Memantine (NMDA receptor antagonist) exhibit inhibitory effects on α-syn cell-to-cell transmission, with advanced clinical trials (NCT03858270) currently evaluating their disease-modifying potential. Concurrent phase I studies targeting mGluR5 receptor (e.g., BMS-984923, NCT06309147) further underscore expanding therapeutic horizons beyond conventional neurotransmitter replacement paradigms (Supplemental Table 2).
However, current evidence in the clinical translation of α-syn-targeted therapies, including monoclonal antibodies (Prasinezumab, Cinpanemab), anti-aggregation vaccines (PD01A), stem cell-based neurorestorative approaches, and other drugs (Supplemental Table 2), has yet to demonstrate statistically significant benefits in modulating neurotransmitter systems,143,144 or has not systematically investigated the effect on neurotransmitters, 145 which could be a key concern in future anti-α-syn drug research. This translation gap likely stems from insufficient mechanistic understanding of α-syn-neurotransmitter crosstalk in human pathophysiology, along with limitations in current biomarker methodologies and few clinical trial designs. Critical challenges persist in establishing causal relationships between α-syn modulation and neurotransmitter dynamics, as well as developing sensitive assays capable of capturing these interactions in clinical populations.
In conclusion, the evolving recognition of α-syn-neurotransmitter interplay as a central PD pathogenesis demands sustained interdisciplinary efforts spanning molecular characterization to therapeutic innovation.
Urgent issues and future perspectives
Since a large number of studies focused on the relationship between α-syn and neurotransmitters, the veil of such a relationship is gradually being lifted, contributing to elucidating the pathogenesis of PD and promoting the diagnosis and treatment of PD. Based on what we have mentioned above, the mutual effects between α-syn and neurotransmitters are obvious, but complex and multifaceted. However, there are still many issues worthy of in-depth investigation in the future.
First, despite numerous studies concentrated on the relationship between α-syn and neurotransmitters, most focused on the catechols. The impact of other neurotransmitters may precede that of catechols, leading to heterogeneous non-motor symptoms in PD. In addition, neurotransmitters do not function in isolation. Thus, the interaction between α-syn and neurotransmitters other than catechols deserves further exploration, as it may help advance early diagnosis of PD and advance the development of multi-targeted therapeutic agents.
Second, most of the outcomes from previous research were derived from in vitro experiments, thereby different results may be attributable to the distinct in vitro models. It is worthwhile to explore whether, and to what extent, these interactions occur in vivo models and in PD patients.
Third, the toxicity of α-syn was significantly influenced by several factors, including its genotypes (wild-type or mutant), PTMs (phosphorylation, acetylation, etc.), composition (monomers, dimers, trimers, oligomers, or fibrils, etc.), morphology of aggregates (pore, spherical, etc.), as well as interaction with other substrates (neurotransmitters modifications, lipids, etc.). Hence, detecting the proportion of different types of α-syn may be important for better understanding PD pathogenesis and monitoring disease progression.
Surprisingly, with advancements in detection techniques, researchers could detect catechols-modified α-syn in the plasma with mass spectrometry, 134 but the structure of α-syn remains unknown. Also, most current detection kits for α-syn can only roughly quantify monomers or oligomers and cannot further classify oligomers precisely. Although the prevalent seed amplification assay has displayed outstanding advantages in early diagnosis as well as differential diagnosis of the PD, this assay could not monitor the progression of PD due to its qualitative nature. Thus, establishing an assay with high sensitivity and specificity that can simultaneously detect and classify distinct forms of α-syn, particularly oligomers, will complement the current assays, which may be beneficial for the PD diagnosis and treatment, as well as provide deeper insight into the specific causative form of α-syn. Further, the question of whether interaction between α-syn and neurotransmitters is neuroprotective or neurotoxic could be answered.
Conclusion
To sum up, in regard to the significant abnormal alterations that occur in α-syn and neurotransmitters in PD, including increased α-syn oligomerization and imbalance in neurotransmitters’ homeostasis (primarily catechols), studies on their pathogenic role and the impact of their interactions in PD’s pathogenesis are progressively deepening. For neurotransmitters, α-syn could strongly suppress the synaptic vesicle trafficking process and exert a series of specific effects on certain neurotransmitters. Conversely, neurotransmitters led by catechols also play an important role in inducing α-syn oligomerization and other effects. Additionally, despite the widely acknowledged synergistic toxic effects of α-syn and neurotransmitters, mainly catechols, several recent studies revealed the presence of a neuroprotective role of their interactions. It is worthwhile to conduct more profound research on interactions between different types of α-syn and neurotransmitters, particularly in vivo, to figure out what proportion and form of interactions contribute to neurotoxic or neuroprotective effects in PD. Finally, it is hoped that the development of multiple biomarker platforms integrating α-syn with neurotransmitters, along with rationally designed multi-target therapeutics validated through large-scale clinical trials, will advance the diagnosis and treatment of PD.
Supplemental Material
sj-docx-1-tan-10.1177_17562864251339895 – Supplemental material for Decoding crosstalk between neurotransmitters and α-synuclein in Parkinson’s disease: pathogenesis and therapeutic implications
Supplemental material, sj-docx-1-tan-10.1177_17562864251339895 for Decoding crosstalk between neurotransmitters and α-synuclein in Parkinson’s disease: pathogenesis and therapeutic implications by Lihua Guan, Liling Lin, Chaochao Ma and Ling Qiu in Therapeutic Advances in Neurological Disorders
Supplemental Material
sj-docx-2-tan-10.1177_17562864251339895 – Supplemental material for Decoding crosstalk between neurotransmitters and α-synuclein in Parkinson’s disease: pathogenesis and therapeutic implications
Supplemental material, sj-docx-2-tan-10.1177_17562864251339895 for Decoding crosstalk between neurotransmitters and α-synuclein in Parkinson’s disease: pathogenesis and therapeutic implications by Lihua Guan, Liling Lin, Chaochao Ma and Ling Qiu in Therapeutic Advances in Neurological Disorders
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.