
Abstract
Parkinson’s disease is a slowly progressive neurodegenerative disorder typically characterized by the loss of dopaminergic neurons within the substantia nigra pars compacta, and the intraneuronal deposition of insoluble protein aggregates chiefly comprised of α-synuclein. Patients experience debilitating symptoms including bradykinesia, rigidity and postural instability. No curative treatment currently exists and therapeutic strategies are restricted to symptomatic treatment only. Over the past decade a class of molecular chaperones called the heat shock proteins has emerged as a potentially promising therapeutic target. Heat shock proteins aid in the folding and refolding of proteins, and target denatured proteins to degradation systems. By targeting heat shock proteins through various means including overexpression and pharmacological enhancement, researchers have shown that α-synuclein aggregation and its associated cytotoxicity can be therapeutically modulated in an array of cell and animal models. This review highlights the relevant progress in this field and discusses the relevance of heat shock proteins as therapeutic modulators of α-synuclein toxicity to the rapidly evolving understanding of Parkinson’s disease pathogenesis.
Introduction to Parkinson’s disease
Parkinson’s disease (PD) is a progressive degenerative movement disorder of the nervous system. Next to Alzheimer’s disease, PD is the second most common neurodegenerative disorder and primarily affects the elderly population with an average onset age of around 60 years. PD also affects the younger population; for example, young onset PD affects around 10% of all PD patients and occurs between the age of 21 and 40 years [Quinn et al. 1987]. The exact cause of PD remains obscure for the majority of PD patients, although around 10% have some genetic/familial cause. Genes encoding α-synuclein, LRRK2, Parkin, DJ1, PINK1, ATP13A2, VPS35, FBXO7, GBA and most recently the EIF4G1 gene have been shown to be implicated in the pathogenesis of and susceptibility to PD, and are reviewed elsewhere [Kitada et al. 1998; Singleton et al. 2003; Chartier-Harlin et al. 2004, 2011; Di Fonzo et al. 2009; Sidransky et al. 2009; Shulman et al. 2011].
Males appear to be at a slightly higher risk for sporadic PD than females, and head trauma, repeated exposure to pesticides, and aging in particular increase the risks of developing PD. It has also been hypothesized that brain inflammation (in particular microglial activation), which is induced by these and other PD risk factors, might play an important early pathogenic role in PD [Rogers et al. 2007]. In addition, secondary PD can be chemically induced by neurotoxins such as rotenone, 6-hydroxydopamine and the mitochondrial toxin, 1-methyl-4-phenyl-1,2-3,6-tetrahydropyridine (MPTP). In fact MPTP induced chronic parkinsonism in four recreational drug users following an unsuccessful attempt to synthesize the opioid analgesic, 1-methyl-4-phenyl-4-propionoxypiperidine (MPPP), and since this event MPTP has become a means of modeling the disease in vivo [Langston et al. 1983; Dauer and Przedborski, 2003].
In addition to early nonmotor symptoms such as loss of smell, constipation and insomnia, PD patients experience an array of debilitating symptoms which usually occur as the disease progresses and include tremor, rigidity, postural instability and bradykinesia. These are a result of neuropathological changes affecting an assortment of neurotransmitter systems within the brain, particularly the dopamine system [Karachi et al. 2010]. The exact cause of these neuropathological changes remains obscure, however.
Current approaches to the symptomatic treatment of PD
To date, PD remains an incurable neurodegenerative disorder. Indeed the pathogenesis of PD is still unclear and research into its molecular mechanisms is ongoing. The cellular degeneration characteristic of PD primarily affects the pigmented dopaminergic neurons of the substantia nigra pars compacta (SNpc), as well as cholinergic neurons such as those of the pedunculopontine nucleus (PPN) in the mesencephalic locomotor region [Karachi et al. 2010]. The primary parkinsonian symptoms are directly linked to a reduction in the production of the monoamine neurotransmitter dopamine (DA) caused by the loss of DA neurons, and the loss of PPN neurons.
Current therapeutic strategies are restricted to symptomatic treatments only [Weintraub et al. 2008]. Of these, the large majority target the dopaminergic neurotransmitter system and aim to replace lost dopaminergic transmission at the striatal synapses, either by increasing DA levels or by stimulating the central DA receptors (Figure 1). Often these strategies may be combined in order to optimize symptomatic treatment. The standard approach to the relief of PD symptoms is

Dopaminergic synapse and dopamine metabolism.
Several other PD treatments have been marketed, such as DA agonists (including pramipexole, ropinirole, bromocriptine, rotigotine and apomorphine) that bind directly to dopaminergic receptors and increase the dopaminergic signaling; but also monoamine oxidase (MAO) inhibitors (selegiline and rasagiline) and cathecol-O-methyl transferase (COMT) inhibitors (entacapone and tolcapone) which decrease DA degradation (Figure 1). Treatments that aim to continuously stimulate dopaminergic neurons, and hence to avoid intermittent stimulation, are reviewed by Sujith and Lane [Sujith and Lane, 2009].
Other treatment options currently available include amantadine and anticholinergic substances, deep brain stimulation or pallidotomy (reviewed elsewhere; [Jankovic and Aguilar, 2008]). These measures aim to manage the symptoms of PD in order to maintain a better quality of life for the patient, at least during the first stages of the disease. However, curable treatments or strategies which are able to slow the progression of PD are urgently needed as none currently exist in the clinic.
α-Synuclein and PD
PD is pathologically characterized by a selective loss of dopaminergic neurons in the SNpc and the presence of abnormal intracellular protein deposits called Lewy bodies (LBs) and Lewy neurites (LNs). As their names indicate, LBs and LNs are proteinaceous cytoplasmic inclusions localized in the cell body and cell neurites of neurons, respectively. They are the defining neuropathological hallmarks of degenerating and perhaps surviving neurons found in the brains of PD patients and the pathologically related disorders such as dementia with Lewy bodies (DLB) [Irizarry et al. 1998; Spillantini et al. 1998a, 1998b; Li et al. 2010]. LBs and LNs can be histologically detected by eosin staining, or by anti-ubiquitin or anti-α-synuclein immunohistochemistry. In PD pathogenesis, endogenous α-synuclein is believed to misfold and oligomerize, triggering a polymerization cascade which ultimately leads to the formation of LBs and LNs; indeed α-synuclein was found to be the major component of these aggregates. In addition to α-synuclein, LBs and LNs contain around 250 other proteins including ubiquitin, p62 and a multitude of molecular chaperones [Spillantini et al. 1997; Kuusisto et al. 2003].
α-Synuclein is a 140 amino acid protein of ambiguous function with a molecular weight of 14.46 kDa, which is encoded by the SNCA gene (Figure 2). The protein contains multiple repeated domains of 11 residues, which display variations of the consensus sequence KTKEGV (depicted in blue, Figure 2a). It is composed of three major regions with a stretch of hydrophobic 12 amino acid residues in the middle which has been shown to be essential for filament assembly during disease pathogenesis [Giasson et al. 2001] (Figure 2). The C-terminus is a highly acidic, proline-rich region containing four putative phosphorylation sites that is randomly coiled with no apparent secondary structure. By contrast, the N-terminal region from residues 1 to 60 was shown to bind to biological membranes by adopting an amphipathic α-helical conformation [Davidson et al. 1998].

α-Synuclein structures and the pathogenic process of polymerization
Although its exact function is still elusive, α-synuclein has been implicated in synaptic transmission and induction of neurotransmitter release. Moreover, other purported functions include vesicular trafficking, neuroplasticity and fatty acid binding [Abeliovich et al. 2000; Cabin et al. 2002; Liu et al. 2004; Yavich et al. 2004]. In addition to its high abundance in the presynaptic terminals of neurons, α-synuclein is also expressed in other areas of the central nervous system (CNS) and in peripheral tissues including muscle and kidney [Ueda et al. 1993; Lavedan et al. 1998].
Interestingly, there is strong evidence that α-synuclein aggregation is an early step in the pathogenesis of PD. For example, duplication or triplication of the number of gene copies of the wildtype form of the α-synuclein gene induces an important genetic risk factor for the development of PD [Singleton et al. 2003; Chartier-Harlin et al. 2004]. Additionally, five point mutations in the N-terminal region of α-synuclein (A30P, E46K, H50Q, G51D and A53T) are responsible for autosomal dominant familial forms of PD (Figure 2) [Polymeropoulos et al. 1997; Kruger et al. 1998; Zarranz et al. 2004; Appel-Cresswell et al. 2013; Kiely et al. 2013]. Moreover, α-synuclein aggregation is also observed in other closely related neurodegenerative disorders termed synucleinopathies. Synucleinopathies include PD and PD with dementia (PDD), but also DLB, pure autonomic failure and multiple system atrophy. In all these diseases α-synuclein forms abnormal intracellular protein aggregates. Taken together, these observations strongly suggest a causative role of α-synuclein in neurotoxicity and a complete understanding of α-synuclein aggregation will be crucial to the development of curative treatments for these diseases.
The exact toxic mechanism(s) associated with pathogenic α-synuclein is somewhat obscure. Interestingly there is an obvious parallel between synucleinopathies and a larger group of diseases which include Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, prion disease and type II diabetes. Indeed the synucleinopathies and these other disorders all belong to a larger group, termed protein-misfolding diseases (PMDs) [Walker and Levine, 2000]. Similarly to synucleinopathies, other PMDs are typically characterized by the anomalous deposition of insoluble aggregates comprised of mutant or misfolded self-proteins, with concurrent cellular degeneration. Protein misfolding is usually a sporadic event or may occur by idiopathic means. In PD, the resultant cellular degeneration that follows α-synuclein misfolding and oligomerization primarily affects the pigmented dopaminergic neurons of the SNpc.
Much debate has surrounded the cause of degeneration in synucleinopathies and other PMDs; a loss of function of the host protein, or a gain of toxic function during the misfolding or oligomerization events [Winklhofer et al. 2008]. Mounting evidence suggests that soluble oligomers formed relatively early during the formation of fibrils are in fact the toxic species, and though the exact mechanism of toxicity remains enigmatic, the pathogenic process may involve multiple systems including the ubiquitin–proteasome system, the autophagy–lysosomal pathway, mitochondrial dysfunction and the unfolded protein response [Greenamyre et al. 1999; Welihinda et al. 1999; Heinitz et al. 2006; Winner et al. 2011; Larson and Lesne, 2012]. For some PMDs, however, a loss of function of a host protein is indeed clear; for example in the case of cystic fibrosis, the cystic fibrosis transmembrane conductance regulator protein (which acts to transport chloride and thiocyanate ions out of epithelial cells to the covering mucus) incurs a loss of function upon misfolding and, as such, has devastating consequences to normal mucus production [Rommens et al. 1989; Knorre et al. 2002].
In the case of PD, α-synuclein appears to be toxic upon overexpression and during misfolding or subsequent oligomerization. In fact protofibrillar α-synuclein was shown to bind and transiently permeabilize synthetic vesicles, and several types of soluble oligomers increased lipid bilayer conductance suggesting that this may be a central mechanism of cytotoxicity in PD and indeed other PMDs [Volles et al. 2001; Lashuel et al. 2002; Kayed et al. 2004]. As α-synuclein (or species thereof) is strongly implicated as the fundamental protein involved in the pathophysiology and pathogenesis of PD, targeting the modulation of α-synuclein and its associated cytotoxicity is a plausible strategy. Indeed, in other closely related PMDs, therapeutic strategies have likewise aimed at targeting the misfolded protein involved. For example, prion diseases are caused by misfolded species of the endogenous prion protein and therapeutic strategies aimed at targeting prions using antibody therapy have been shown to prevent disease progression in prion-infected mice and have cured prion-infected neuroblastoma cells [White et al. 2003; Jones et al. 2010]. However targeting endogenous proteins, or variants thereof, may have unpredictable side effects in humans and may not prevent cytotoxicity as multiple species (and therefore multiple targets) of pathogenic misfolded proteins each with different secondary structures may exist in the diseased-state [Laganowsky et al. 2012]. Thus modulating toxicity by aiding refolding or promoting degradation of misfolded proteins such as α-synuclein might be a more feasible and physiologically relevant approach. Opportunely, heat shock proteins perform these functions under physiologically normal conditions and under conditions of stress [Schmitt et al. 2007]; as such targeting these proteins is a promising means of modulating α-synuclein toxicity in PD and other synucleinopathies.
Protein folding and heat shock proteins
Heat shock proteins (HSPs) are a class of molecular chaperones intimately involved in the folding, unfolding and refolding of proteins which function in cohort with cochaperones. Following (or during) protein translation, polypeptide chains fold with the aid of these chaperones into functional native structures as determined by their amino acid sequences [Anfinsen, 1973; Kolb et al. 1995; Hardesty and Kramer, 2001]. Abnormal protein-folding is a common event during this process; however, the production of redundant or harmful nonnative structures is usually prevented by the presence of such molecular chaperones and other quality control mechanisms which often target denatured proteins to the proteasome or lysosome for degradation [Hightower, 1991; Sherman and Goldberg, 2001; Hartl and Hayer-Hartl, 2002; Sitia and Braakman, 2003; Shorter and Lindquist, 2004]. Under conditions of stress, HSPs and their cochaperones are upregulated to help prevent misfolding of endogenous proteins; when such quality control mechanisms fail, however, the resultant misfolded proteins or oligomeric species thereof may become pathogenic [Winklhofer et al. 2008].
HSPs are a large group of chaperone proteins existing within eukaryotic cells that are classed according to their molecular weight. The two major mammalian chaperone systems are comprised of proteins of around 70 kDa and 90 kDa [heat shock protein 70 family (Hsp70) and Hsp90 family, respectively]. HSPs were identified in 1974 following earlier experiments which noted that temperature increases induce puffing patterns in the polytene chromosomes of Drosophila [Ritossa, 1962; Tissieres et al. 1974]. In fact the HSPs are not limited to induction upon heat stress but are also upregulated during other physiological stresses, including stress induced by toxins and starvation for example [Santoro, 2000]. Furthermore, some members of the HSP families are expressed constitutively (such as heat shock cognate 70; Hsc70) and are intimately supported by the cochaperones which help to regulate the function of chaperones [Erbse et al. 2004; Kalia et al. 2010].
The mechanisms by which HSPs function may vary between HSP families; Hsp70 members contain a binding site at the C-terminus to which exposed hydrophobic regions of denatured or nonnative proteins bind, and an ATPase domain in the N-terminus which facilitates the hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP). This hydrolysis of ATP to ADP at the N-terminus facilitates conformational changes at the C-terminus so that the bound, denatured protein is stabilized in a so-called ‘hold’ conformation. It is during subsequent cycles in which ADP is released in exchange for ATP that the denatured protein is believed to be refolded [Goloubinoff and De Los Rios, 2007; Kalia et al. 2010].
Due to their ability to refold denatured proteins, or to target denatured proteins to the degradation systems, HSPs have been viewed as potential candidates for the treatment of diseases in which such denatured proteins are closely associated with disease pathogenesis. Indeed in PD, strategies which target the HSPs are actively being explored as a plausible means of modulating α-synuclein associated toxicity (Figure 3) [Luo et al. 2007; Kalia et al. 2010].

Activation of heat shock transcription factor-1 by Hsp90-inhibition and modulation of α-synuclein aggregation by heat shock proteins.
The emergence of HSPs as therapeutic targets
The naturally occurring antibiotic geldanamycin (GA) (a benzoquinone ansamycin) was found to selectively bind members of the Hsp90 family of molecular chaperones [Whitesell et al. 1994]. Like Hsp70, Hsp90 is an ATP-cleaving chaperone intimately involved with the 26S proteasome that binds unfolded/misfolded proteins via its exposed hydrophobic regions at the C-terminus [Imai et al. 2003]. Hsp90 plays an integral role in the cellular transcriptional response to endoplasmic reticulum (ER) stress because it stabilizes the transmembrane kinase ER stress sensors IRE1α and PERK, which, upon activation, initiate downstream events of the unfolded protein response [Marcu et al. 2002; Schroder and Kaufman, 2006]. Inhibition of Hsp90 by GA first emerged as an antiproliferative strategy in the development of cancer therapeutics because Hsp90 also stabilizes a range of proteins involved in the regulation of the cell cycle, cell survival and oncogenesis (reviewed elsewhere [Miyata, 2005]). As a consequence of inhibiting Hsp90, GA induces a cellular heat shock response through indirect activation of its central mediator the heat shock transcription factor 1 (HSF-1) (Figure 3) [Zou et al. 1998]. HSF-1 is deacetylated and binds the Hsp70 promoter [Westerheide et al. 2009]. Indeed systemic administration of GA and other Hsp90-binding agents induced a heat shock response, evident by a marked increase in cytosolic levels of Hsp70 (Hsp72) in normal mouse tissues and human tumor xenografts; furthermore, GA was also shown to induce a heat shock response in an in vitro model of Huntington’s disease [Bagatell et al. 2000; Sittler et al. 2001].
During erroneous protein folding, the Hsp70 family of molecular chaperones is upregulated in consortium with other HSPs (including Hsp40 and Hsp104) in an attempt to rescue misfolded or aggregated proteins [Glover and Lindquist, 1998]. In fact the neuronal toxicity associated with abnormally folded proteins has been successfully suppressed by HSP modulation in numerous models including via the overexpression of Hsp70 and Hsp40. For example, the polyglutamine diseases are a group of neurodegenerative disorders caused by a pathological repetition of polyglutamine units sharing many characteristics with other PMDs including synucleinopathies. The diseases are characterized by the deposition of ubiquitinated nuclear inclusions (NIs) which, like LBs and LNs of synucleinopathies, sequester an array of proteins including molecular chaperones. Overexpression of Hsp40 in HeLa cells transiently expressing a mutant polyglutamine repeat in a model of spinocerebellar ataxia type 1 decreased ataxin-1 NIs, and for the first time implicated protein misfolding in the polyglutamine disorders [Cummings et al. 1998]. Following this, directed expression of Hsp70 in a Drosophila model of polyglutamine disease was shown to suppress neurodegeneration [Warrick et al. 1999]. Interestingly, although it was hypothesized that induction of Hsp70 might disaggregate NIs, no observable effect was found on their formation despite suppression of neurodegeneration, and thus it was concluded that polyglutamine toxicity could be dissociated from the presence of NIs in this model. In a later study it was shown that Hsp70 and Hsp40 (HDJ1) actually suppress polyglutamine disease in the Drosophila model in a synergistic manner seemingly by altering the solubility of the pathological proteins [Chan et al. 2000]. In support, Hsp40 was postulated to be a cochaperone for Hsp70 as it binds to, and stimulates, the ATPase activity of Hsp70 [Cyr et al. 1994]. Furthermore Hsp40 (HDJ2) was shown to suppress aggregate formation in a model of spinal bulbar muscular atrophy caused by a polyglutamine expansion in the androgen receptor, suggesting a feasible means of therapeutic intervention by HSPs in the polyglutamine diseases [Stenoien et al. 1999].
In light of the similarities between polyglutamine diseases and the synucleinopathies, directed expression of Hsp70 was next investigated in a Drosophila model of PD, and indeed was found to prevent dopaminergic neuronal loss associated with expression of α-synuclein in the fly [Feany and Bender, 2000; Auluck et al. 2002]. Furthermore, co-expression of Hsp40 (HDJ1) and Hsp70 suppressed α-synuclein aggregation in a cell culture model [Mclean et al. 2002]. In providing evidence for a beneficial effect of one modifier (Hsp70) in two distinct models of PMDs, the authors of these studies also inadvertently helped to elucidate the paradigm that PMDs might have a unifying mechanism of pathogenesis [Dobson, 2001, 2004], and thus a unifying therapeutic approach may be applicable for many, if not all, PMDs. Consequently, HSPs appeared as a potentially novel treatment for the synucleinopathies and other PMDs, and inhibition of Hsp90 was identified as a means of modifying their expression.
The therapeutic effects of Hsp70 upregulation
Since HSPs emerged as potentially therapeutic modifiers of cytotoxicity in PMDs, mounting data suggest that upregulating Hsp70, either by directed overexpression or indirectly via inhibition of Hsp90 (Figure 3, Table 1) might be the most favorable approach. In support, polymorphisms that affect Hsp70 activation were found to be more frequent in PD than controls [Wu et al. 2004], and numerous studies investigating Hsp70 as a target to modulate α-synuclein toxicity have produced encouraging and supporting data. For example, crossing mice which overexpress Hsp70 with α-synuclein transgenic mice led to a marked reduction in abnormal Triton X-100 insoluble and high molecular weight (HMW) α-synuclein species, which were similarly present in the brains of patients with DLB, but not controls [Klucken et al. 2004]. Total α-synuclein protein levels did not differ between α-synuclein transgenic mice and α-synuclein transgenic mice crossed with mice overexpressing Hsp70, however, despite a reduction in the amount of detergent-insoluble HMW species; thus Hsp70 appears to specifically exert its effects on the abnormal species by influencing solubility and the aggregation process. In agreement, corresponding in vitro studies demonstrated that overexpression of Hsp70 reduced the Triton X-100 insoluble α-synuclein species. These data support the paradigm that overexpression of Hsp70 might be beneficial in human disease because the experimental models described closely resemble the in situ pathophysiology of synucleinopathies such as DLB and PD.
Hsp70 implication in α-synuclein polymerization and neurotoxicity.
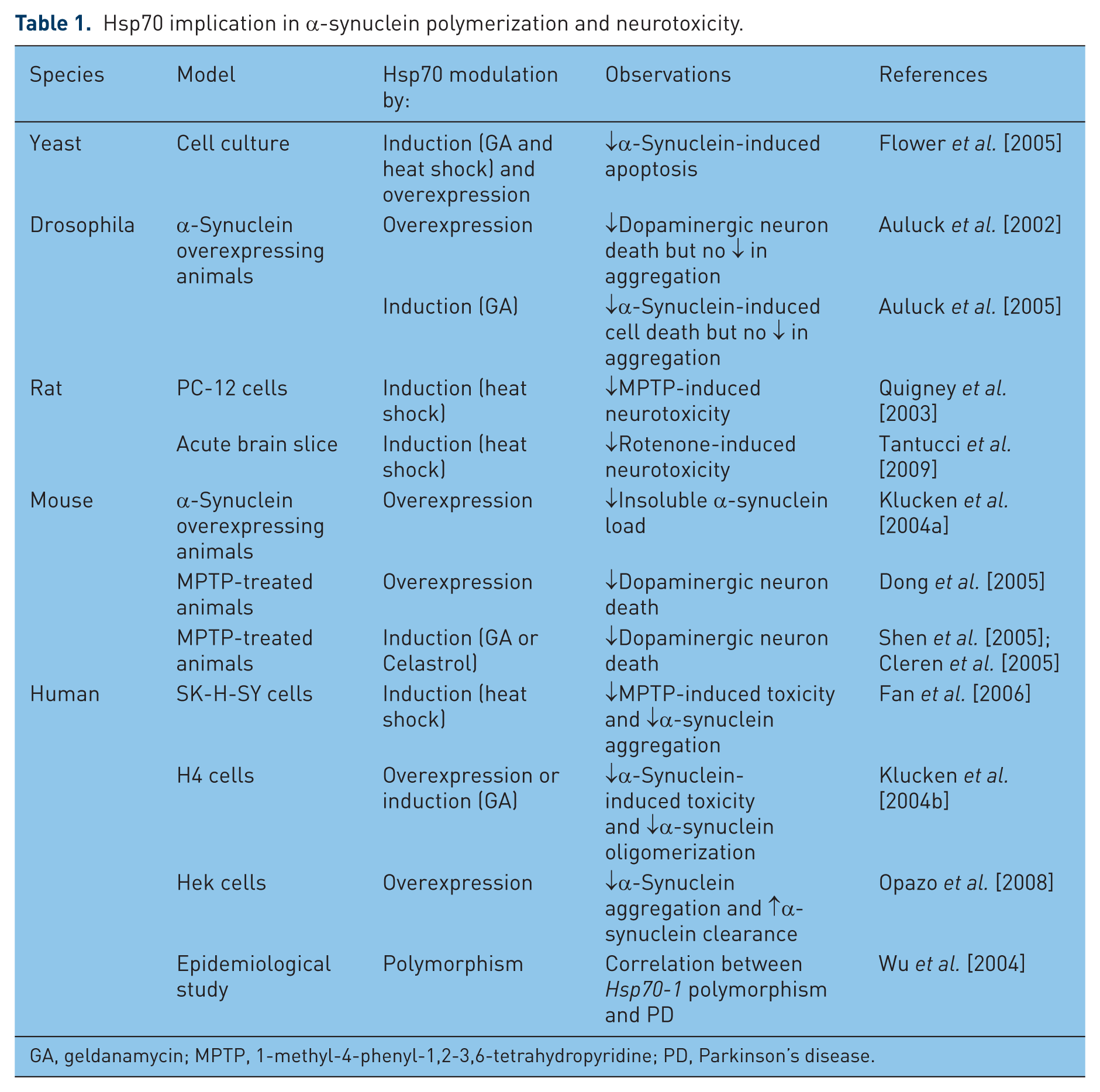
GA, geldanamycin; MPTP, 1-methyl-4-phenyl-1,2-3,6-tetrahydropyridine; PD, Parkinson’s disease.
Influencing the misfolding and subsequent aggregation of α-synuclein might prevent further cytotoxicity and destruction of vulnerable DA neurons in the basal ganglia. In support, Hsp70 gene therapy which used recombinant adenoassociated virus (rAAV) in a MPTP mouse model of idiopathic PD was also shown to be beneficial; indeed Hsp70 protected the dopaminergic system against neuronal loss and MPTP-induced apoptosis, and also protected against the associated decline in DA levels [Dong et al. 2005]. However, such overexpression of Hsp70 by genetic manipulation in human patients is not currently a feasible approach, especially due to unpredictable side effects including unexpected immune responses such as those recently reported in human AAV clinical trials [Mingozzi and High, 2011]. Furthermore, delivery of such viral vectors is restricted to the site of administration, which is not optimal in the case of brain diseases affecting multiple and widespread areas of the brain. Currently, attempts are being made to overcome this caveat [Iwata et al. 2013]. Manipulating Hsp70 by other means is achievable, however, and pharmacological induction of the heat shock response might be a more beneficial strategy. Indeed, Hsp90-inhibitors were shown to maintain chaperone induction in an organotypic slice culture assay for at least 3 weeks and altered the detergent-soluble properties of polyglutamine aggregates; on the contrary, overexpressing Hsp70 in a mouse model of Huntington’s disease had no effect on the detergent solubility of aggregates and delayed aggregate formation by just one week [Hay et al. 2004].
In light of the literature highlighting pharmaceutical Hsp90-inhibiting compounds as novel and beneficial modulators of the heat shock response, GA was further investigated as an inducer of Hsp70 for the treatment of PD. Treatment with GA was found to induce Hsp70 (but not Hsp40) in H4 neuroglioma cells, and cells pretreated with 200nM of GA 24 hours prior to cotransfection with α-synuclein and synphilin-1 (which leads to the development of LB-like inclusions in 50% of cells [Engelender et al. 1999; Mclean et al. 2001, 2002]) led to a significant decrease in the number of cells with LB-like inclusions compared with mock-treated cells [Mclean et al. 2004]. Importantly, this pretreatment prevented α-synuclein induced toxicity; thus upregulating Hsp70 via Hsp90-inhibitors might be an effective means of modulating α-synuclein cytotoxicity in the early phases of human disease (Figure 3). In contrast to pretreatment, however, treatment with GA at the time of transfection or 24 hours post-transfection failed to have any effect; thus Hsp90-inhibition by GA may not be beneficial once neurotoxicity is already established in the brains of PD patients. Indeed there is an urgent need for new and improved early diagnostic markers that might diagnose PD patients before the clinical disease progresses from peripheral to seemingly irreversible nervous symptoms.
HSP mechanisms of action
The mechanisms by which Hsp90 inhibition suppress neurodegeneration in a Drosophila model of PD were investigated following initial studies in the fly, which demonstrated that GA induces Hsp70 in a concentration-dependent manner [Auluck et al. 2002]. Intriguingly it was found that a concentration of GA of 3 µg/ml was sufficient to protect DA neurons against α-synuclein-associated toxicity but was not sufficient to induce a marked increase in Hsp70 [Auluck et al. 2005]. Thus it was hypothesized that the therapeutic effects of Hsp90-inhibition by GA might be due to a general enhancement of the stress response rather than the upregulation of specific HSPs.
Investigative studies revealed that abolishing the HSF gene decreases induction of Hsp70, and GA treatment in mutant HSF flies does not protect against α-synuclein toxicity; thus in the Drosophila model, the neuroprotective effects of Hsp90-inhibition by GA require HSF activity [Auluck et al. 2005]. The authors concluded that Hsp90-inhibitors, such as GA, protect against α-synuclein toxicity through mobilization of the stress response and through increased Hsp70 levels. Given that Hsp70 was not elevated at lower doses but DA neurons were protected, it would have been valuable to determine if Hsp90-inhibition by GA could protect against α-synuclein toxicity in Hsp70 knockout or Hsp70 mutant flies. This was not investigated; however, other pathways which are modulated by GA (such as a decrease in Ras signaling via inhibition of Hsp90) were shown to have no effect on α-synuclein toxicity, and interestingly GA appeared to elevate chaperone levels in stressed cells only [Auluck et al. 2005]. Intriguingly, the same study showed that treatment with GA protected neurons from toxicity despite continued presence of LB-like inclusions, and in contrast, inhibition of HSF activity in fact reduced LN-like pathology in the brains of α-synuclein-expressing Drosophila, but was not neuroprotective. This might suggest that the formation of LBs or LNs is a protective mechanism whereby the soluble toxic species is sequestered to the aggregate in an insoluble and inert form. Indeed the authors demonstrated that Hsp90-inhibition by GA or direct Hsp70 overexpression resulted in an increase in the amounts of insoluble α-synuclein but a decrease in α-synuclein toxicity [Auluck et al. 2005]. This is in agreement with Chan and colleagues who showed that Hsp70 and Hsp40 act in a synergistic manner to alter the solubility of pathological proteins [Chan et al. 2000]. Interestingly however, we found that introducing Hsp70 into an in vivo model of α-synuclein overexpression conversely led to a significant reduction in the detergent-insoluble α-synuclein species [Klucken et al. 2004]. Furthermore, Hsp70 was shown to preferentially bind prefibrillar species associated with cellular toxicity and strongly inhibited α-synuclein fibril formation [Dedmon et al. 2005]. Perhaps therefore Hsp70 is able to target both soluble and insoluble nonnative α-synuclein species. Nonetheless, soluble oligomers are thought to be the toxic species in the pathogenesis of most, if not all PMDs; in contrast, fibril formation indeed appears to be an attempt to sequester oligomer-associated toxicity (reviewed elsewhere [Ferreira et al. 2007]).
In addition to refolding and targeting denatured proteins to degradation systems, some HSPs also have specialized functions which may be exploited to reduce α-synuclein toxicity. For example GRP78/BiP, which is a member of the Hsp70 family, plays an integral role in the unfolded protein response pathway. Indeed when a misfolded protein is bound by GRP78/BiP in the ER, three local stress receptors (namely PERK, IRE1 and ATF6) are dissociated from GRP78/BiP and become activated. Subsequently, downstream events are triggered which lead to apoptosis and the global attenuation of protein translation [Ron and Walter, 2007]. Co-injection of rAAV-GRP78/BiP and rAAV–α-synuclein was shown to reduce α-synuclein neurotoxicity in a rat model of PD by downregulating ER stress mediators including downstream players such as the pro-apoptotic transcription factor ATF4 [Gorbatyuk et al. 2012]. Thus in addition to directly targeting denatured proteins, HSPs also exert therapeutic effects by modulating pathways integrally involved in the maintenance and rescue of normal protein homeostasis.
Other chaperone-like proteins as modulators of α-synuclein toxicity
Hsp70 is the most studied molecular chaperone in regard to α-synuclein aggregation; indeed the effects of Hsp70 upregulation by varying strategies have been investigated in a range of models (Table 1). However, other molecular chaperones have also been shown to affect α-synuclein aggregation, and as much, might represent alternative targets. Moreover Hsp27, Hsp40 (HDJ1; HDJ2), Hsp60, Hsp90, Hsp110, 14-3-3, Hsc70 and αB-crystallin were all shown to be sequestrated in LBs [Mclean et al. 2002; Leverenz et al. 2007]. In addition TorsinA, HDJ1, HDJ2, Hsp27, and to a lesser extent αB-crystallin, were all shown to reduce α-synuclein-induced neurotoxicity in cell culture models [Mclean et al. 2002; Outeiro et al. 2006]. Overexpressing Hsp27 in vitro was shown to be a more favorable approach than overexpressing Hsp70, because Hsp27 had a broader and more potent protective effect against wildtype α-synuclein, and additionally protected against toxicity associated with mutant α-synuclein [Zourlidou et al. 2004]. Interestingly Hsp27 was also shown to suppress toxicity in the polyglutamine diseases [Wyttenbach et al. 2002]. More recently, Bruinsma and colleagues compared in vitro the small HSPs αB-crystallin, Hsp20, Hsp27, HspB8 and HspB2B3 for their ability to interact with α-synuclein, and observed that all chaperones investigated were able to inhibit mature fibril formation to varying extents [Bruinsma et al. 2011]. Indeed, in addition to these and numerous other molecular chaperones, the BAG domain, TRP domain and DNAJ domain containing cochaperones such as Bag1-6, CHIP, Hop and Hip may represent alternative targets for the modulation of α-synuclein toxicity [Mayer and Bukau, 2005]. Interestingly Hip, which is the Hsp70-interacting protein, was found to be consistently underexpressed in PD patients in the early stages of the disease [Scherzer et al. 2007]. Furthermore, Hsp70 is depleted in PD by its aggregation upon interaction with α-synuclein; however, this aggregation can be prevented in vitro by Hip [Roodveldt et al. 2009]. Thus, overexpression of Hip as a means of preventing Hsp70 depletion may be a novel therapeutic strategy in PD.
Pertinently α-synuclein is also believed to function as a cochaperone. Indeed α-synuclein was shown to have striking homology with the 14-3-3 chaperone, which is elevated in prion disease and other CNS assaults including cerebrovascular accident, and like 14-3-3, α-synuclein binds BAD and protein kinase C, modulating their functions [Ostrerova et al. 1999]. Further, the cochaperone function of cysteine-string protein-alpha (CSPα) is essential for neuronal survival, but overexpression of α-synuclein in CSPα knockout mice abolishes the lethality of the deletion by ameliorating the inhibition of SNARE complex assembly, which is required for the release of neurotransmitters at the presynaptic terminals [Chandra et al. 2005]. Taken together, these data indicate that α-synuclein may indeed function as a cochaperone and add weight to the hypothesis that α-synuclein pathogenesis might be alleviated by modulation or enhancement of chaperone activity.
CHIP
The carboxyl terminus of Hsp70-interacting protein (CHIP) was identified as a cochaperone of Hsp70 [Ballinger et al. 1999]. Such cochaperones assist chaperones by regulating their functions. Indeed, via its interaction with HSPs, CHIP was shown to mediate degradation of various misfolded proteins involved in the pathogenesis of Alzheimer’s disease, PD and other PMDs [Miller et al. 2005; Shin et al. 2005; Al-Ramahi et al. 2006; Adachi et al. 2007]. CHIP was also found to be a component of LBs in human brain, where it was shown to colocalize with α-synuclein and Hsp70 [Shin et al. 2005].
CHIP acts as a link between the molecular chaperone system and the protein degradation systems. Indeed, CHIP was shown to mediate α-synuclein degradation by two discrete mechanisms: the proteasomal degradation pathway and the lysosomal degradation pathway [Shin et al. 2005]. CHIP is composed of three major domains: an amino-terminal three tandem tetratricopeptide repeat (TPR) domain, a highly charged central domain, and a carboxyl-terminal U-box domain. The TPR domain links CHIP to Hsp70 and Hsp90, and the U-box domain confers the E3 ubiquitin ligase function of CHIP [Jiang et al. 2001]. We found that the TPR domain of CHIP is critical for proteasomal degradation and that the UPR domain is sufficient to direct α-synuclein to the lysosomal degradation pathway [Shin et al. 2005]. Thus, CHIP might be utilized as a therapeutic modulator of α-synuclein toxicity via its upregulation, by promoting the degradation of toxic α-synuclein. Accordingly, we investigated the effects of CHIP overexpression in an in vitro model of α-synuclein inclusion formation. Indeed, CHIP overexpression was found to inhibit α-synuclein inclusion formation and also reduced α-synuclein protein levels in human H4 neuroglioma cells transiently expressing α-synuclein [Shin et al. 2005]. Furthermore, using a novel bimolecular fluorescence complementation assay under conditions where oligomers were expressed as less stabilized or as more stabilized, CHIP was shown to selectively target the toxic α-synuclein oligomers for degradation [Tetzlaff et al. 2008].
Furthermore, CHIP was shown to ubiquitinylate α-synuclein [Kalia et al. 2011]. Ubiquitinylation is an integral step in the degradation of proteins targeted to the ubiquitin-proteasome proteolytic pathway [Glickman and Ciechanover, 2002]. Thus, CHIP also appears to target α-synuclein to this degradation pathway in an attempt to reduce the toxic load of misfolded α-synuclein. As such, overexpression or upregulation of CHIP might represent a promising therapeutic strategy in PD.
Other means of upregulating Hsp70
Other means of targeting the HSPs include indirect strategies such as targeting HSF-1. Indeed the transcription of Hsp70 is initiated when HSF-1 binds the Hsp70 promoter; this occurs following HSF-1-deacetlyation by the sirtuin, SIRT1 [Westerheide et al. 2009]. As such modulation of SIRT1 might provide a means of modulating Hsp70. Interestingly SIRT1 deletion was shown to enhance levels of acetylated-tau and pathogenic forms of phosphorylated tau involved in the pathogenesis of PMDs, including Alzheimer’s disease and PD [Min et al. 2010]. Furthermore, SIRT1 overexpression in a mouse model of PD was shown to protect against α-synuclein aggregation by upregulating Hsp70 [Donmez et al. 2012]. Interestingly sirtuins can be activated in human patients by resveratrol, a natural plant phenol which is reviewed elsewhere [Alcain and Villalba, 2009; Mai et al. 2009].
Circumventing the blood–brain barrier
A major obstacle to the development of therapies which are directed at targets within the CNS is the inability of most therapeutic compounds to cross the blood–brain barrier (BBB). Indeed, the BBB is both an anatomical and physiological barricade that prevents the access of most peripherally administered drugs to the CNS (reviewed elsewhere [Pardridge, 2005]). Despite promising studies which implicate GA as a promising candidate for therapeutic intervention in PD via induction of the heat shock response, GA does not cross the BBB.
Identifying structurally related analogues to such potentially therapeutic compounds is a common means to circumvent this problem. However, structurally related compounds to GA, including 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), were found to be suboptimal and human clinical trials in the cancer field presented with varying problems including drug-induced liver injury [Waza et al. 2006] (discussed elsewhere [Chiosis and Tao, 2006]). In light of this, a group of chemically dissimilar Hsp90-inhibiting compounds was screened; the compounds belong to the SNX-2112 drug class which was found to be a promising alternative in the cancer field [Chandarlapaty et al. 2008; Okawa et al. 2009]. Several SNX compounds were found to prevent α-synuclein oligomerization in an in vitro model of dimerization [Remy and Michnick, 2006; Putcha et al. 2010], and in agreement with previous work which investigated Hsp90-inhibition and/or Hsp70 overexpression as a means of modulating α-synuclein toxicity, several members of the SNX drug class reduced HMW oligomeric α-synuclein species and protected against α-synuclein-associated cytotoxicity [Putcha et al. 2010]. Interestingly, compound SNX-3113 appeared to shift the HMW α-synuclein species to monomers, suggesting that it possesses unfolding or refolding capabilities. All the Hsp90 inhibitors increased Hsp70 levels, and a lead compound (SNX-0723) reduced α-synuclein oligomerization by up to 83% and prevented α-synuclein toxicity by up to 40% in vitro. Crucially, SNX-0723 also showed rat brain concentrations at therapeutically relevant concentrations at 6 hours following oral dosing and induced Hsp70 in the brain. Thus SNX-0723 might represent a feasible means of modulating α-synuclein toxicity in the brains of PD patients, as it crossed the BBB following oral administration in an in vivo model. More recently we have generated unpublished data which show that chronic treatment with two leading SNX compounds (SNX-0723 and SNX-9114) appear to rescue α-synuclein induced striatal DA depletion compared with vehicle controls.
In conclusion, given these recent novel data and the large quantity of data previously generated implicating Hsp90 inhibition as a promising strategy for the treatment of PD, further investigations of this SNX drug class are indeed warranted. Particular effort should now be given to advancing the in vivo studies and to translating the data from the bench to the clinic.
Other attempts to bypass the BBB using HSP-modulating strategies include the generation of a novel fusion protein that contains a Hsp70 sequence tagged with a cell-penetrating peptide. In a PD animal model, this modified version of Hsp70 was shown to cross both cell membranes and the BBB, and efficiently protected dopaminergic neurons from α-synuclein toxicity [Nagel et al. 2008]. Given that HSPs directly bind and ‘hold’ denatured proteins (discussed previously), crossing the cell membrane may be a therapeutic advantage for the refolding or unfolding of intracellular α-synuclein aggregates which may be inaccessible to extracellular peptides. These and other novel strategies continue to push the boundaries currently limiting the development of effective therapeutic strategies in the field of PD therapeutics.
Emerging paradigms of α-synuclein toxicity and the relevance to HSPs
Since the report emerged that embryonic tissue grafted into the brains of PD patients later developed LB pathology, the idea that extracellular α-synuclein might have been transported to the grafts and thus might play an integral role in the pathogenesis of PD has gained much momentum [Kordower et al. 2008]. Accordingly, it is hypothesized that cell–cell transmission of PD pathology might occur via extracellular release of α-synuclein; in support, α-synuclein was shown to be present in the culture medium of neuroblastoma cells and primary neurons [Lee et al. 2005]. In fact, extracellular α-synuclein was shown to be resecreted out of neurons by a regulator of the recycling endosome (rab11a) [Liu et al. 2009]. Moreover, Hsp90 was shown to interact with rab11a during this process and Hsp90 -inhibition prevented the resecretion of extracellular α-synuclein and attenuated the toxicity associated with extracellular α-synuclein. Thus in addition to inducing a heat shock response and upregulating Hsp70, inhibiting Hsp90 might additionally prevent the release of extracellular α-synuclein and thus prevent or decelerate propagation of α-synuclein pathology. This exciting report offered a remarkable insight into the currently evolving understanding of PD pathogenesis and provided further mechanistic insights into the therapeutic effects of targeting Hsp90 as a means of modulating α-synuclein toxicity.
Following this, Danzer and colleagues next demonstrated the presence of α-synuclein oligomers in the extracellular space using a protein complementation assay in which α-synuclein oligomerization can be monitored [Tetzlaff et al. 2008; Putcha et al. 2010; Danzer et al. 2011]. Furthermore, the uptake of α-synuclein oligomers by neurons and their subsequent axonal transport to the soma was also verified. Interestingly, secreted α-synuclein was found to be toxic to neighboring cells by activation of caspase-3/7 [Danzer et al. 2011]. Co-expression of Hsp70 influenced the formation of extracellular oligomers; indeed α-synuclein oligomer formation in the culture medium of cells expressing Hsp70 was reduced by 64%. More striking, however, was that inhibition of Hsp90 reduced extracellular α-synuclein oligomer formation by 84%, which again suggests that the pharmacological upregulation of Hsp70 via Hsp90 inhibition is a more attractive approach than directed expression of Hsp70. This is presumably due to a dual effect whereby inhibition of Hsp90 induces Hsp70, and thus promotes refolding and disaggregation of α-synuclein oligomers, but also prevents the release of extracellular α-synuclein via a reduced availability of Hsp90 for interaction with rab11a [Liu et al. 2009; Danzer et al. 2011].
Final remarks
Targeting the HSPs, and indeed other molecular chaperones, has emerged as a promising means of modulating α-synuclein toxicity in PD and other PMDs. The current understanding of the pathogenesis of PMDs suggests that the fundamental process involved is the pathological misfolding and subsequent oligomerization of otherwise endogenously native proteins. Given that the HSPs are intimately involved in the normal folding of proteins, are upregulated under conditions of stress, and target denatured proteins to degradation systems, targeting members of this family to enhance the clearance or refolding of misfolded proteins seems a most plausible strategy for the development of an effective therapy.
At present, the most defined approach is the upregulation of Hsp70 by overexpression, or by Hsp90 inhibition. Pharmacological means of achieving Hsp70 induction appear to be most favorable; indeed direct overexpression of Hsp70 such as by the use of AAV vectors, for example, is currently unpredictable. However, the future of effective neurodegenerative therapies may indeed rest in the improvement of currently available gene delivery systems.
Importantly, new and improved diagnostic markers are urgently needed. Accurately diagnosing PD patients in the early stages of disease might be of paramount importance in preventing further neuronal loss. For example, targeting HSPs to modulate α-synuclein toxicity might indeed prevent further neurodegeneration in PD patients, but is unlikely to reverse neuronal loss already present in the SNpc or other areas of the brain.
Excitingly, as our understanding of PD pathogenesis has recently progressed, so too has our understanding of how HSPs might act therapeutically. Indeed, enhancing the heat shock response appears to enhance a range of homeostatic pathways involved in protein folding and degradation, and also appears to target extracellular α-synuclein, a mechanism which may help to prevent the propagation of PD pathology.
Finally, in light of the fundamental similarities between PD and other synucleinopathies and PMDs, targeting the HSPs to modulate α-synuclein toxicity in PD is also likely to be applicable to other protein misfolding disorders including Alzheimer’s disease, which is the most common neurodegenerative disorder of the 21st century.
Footnotes
Acknowledgements
The authors would like to thank Marion Delenclos for valuable input into the structure of this manuscript. The authors would also like to thank Thomas W. Baine V and Ann-Marie T. Baine for proofreading this manuscript.
Funding
This work was financially supported by NIH NS063963, and NS073740 (P.J.M.).
Conflict of interest statement
The authors declare no conflict of interest in preparing this article.