
Abstract
Currently, the diagnosis of Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and Lewy body dementia (DLB) relies on clinical symptoms, with Lewy body (LB) pathology serving as the gold standard. The co-pathology associated with Alzheimer’s disease (AD) contributes to the clinical heterogeneity and rapid progression seen in Lewy body disorders (LBD). The AT(N) classification system may help identify the distinct biochemical, neuro-radiological, and clinical characteristics of both pure LB and PD. Recent advancements in biomarkers have improved the precise identification of pathological α-synuclein (i.e., misfolded and aggregated) in cerebrospinal fluid (CSF) through the seed amplification assay. Consequently, the Neuronal α-synuclein Integrated Staging System (NSD-ISS) has reclassified PD as a neuronal α-synuclein disease, rather than just a clinical syndrome. Although some debate the necessity of a biological definition for clinical diagnosis, biomarker-based systems continue to serve as diagnostic tools for these disorders. This narrative review will explain the definitions of the AT(N) and NSD-ISS systems and provide an updated list of research that supports the proposed biological definitions and staging systems. Additionally, it will discuss how the combination of LB-AD pathology, along with the neuronal concept of the disease, significantly influences the clinical phenotype, progression, and overall prognosis of PD. Finally, this review will overview current advancements in blood-based AD biomarkers that could facilitate faster screening of LBD patients for AD co-pathologies, thereby enhancing the diagnostic sensitivity of LBD-AD and its potential for prognostic, research, and diagnostic applications.
Keywords
Introduction
The broad category of Lewy body disease (LBD) includes three primary disease types: Parkinson’s disease (PD), Parkinson’s disease with dementia (PDD), and dementia with Lewy bodies (DLB). 1 A typical observation in LBD is the presence of intraneuronal inclusions of the presynaptic protein α-synuclein (α-syn), which are known as Lewy bodies and Lewy neurites, collectively referred to as Lewy pathology. One clinical distinction between PDD and DLB is the “one-year rule,” which is somewhat arbitrary and does not necessarily reflect pathological overlaps and differences among the LBD subgroups. To address this, a biological staging paradigm, based on biomarker technologies, may facilitate the selection of regulatory-acceptable trial populations across the LBD continuum, enable stage-appropriate endpoint selection, and accelerate therapy testing. For example, the FDA-approved medications lecanemab and donanemab, which aim to reduce clinical symptoms, have recently been validated by the biological classification of Alzheimer’s disease (AD) in live humans, alongside a grading system based on biomarker presence and symptom severity.2,3 Two new biological classification systems for PD have also been published, attempting to replicate the neuropathological changes that occur over a person’s lifetime.4,5 Individuals with PD are further categorized based on their clinical status (i.e., whether or not they exhibit PD-related clinical symptoms), the presence or absence of synucleinopathy (S status), persistent neurodegeneration (N status), or polymorphic gene variation (G status), within the SynNeurGe system. 4 The neuronal α-synuclein disease integrated staging system (NSD-ISS), presented by Simuni et al., 5 incorporates the identification of genetic (G) risk status, dopaminergic neuronal dysfunction (D anchor), and the detection of abnormal α-syn species (S anchor). Nonprofit organizations promote discussion and contributions toward a biological stage system for developing treatment for PD and DLB, facilitating this progress.
The underlying pathological background is strongly associated with the clinical presentation of patients with LBD, particularly regarding cognitive deterioration.1,6–8 At least one additional neuropathological characteristic is found in 70%–80% of LBD cases, often including pathological signs of AD, such as neurofibrillary tau tangles (NFTs) and amyloid-β (Aβ) plaques. Three sets of biomarkers can be distinguished based on their pathological correlates: tau pathology (T), neurodegeneration (N), and Aβ deposition (A).9–11 Distinct pathologic stages of AD are indicated by biomarker patterns such as A+T−(N)− and A+T−(N)+. 12 The AT(N) system is adaptable, allowing for the integration of new biomarkers into the three AT(N) groups and the inclusion of additional biomarker categories as required. Consequently, the revised 2024 criteria introduce three additional biomarker categories: vascular brain injury (V), alpha-synucleinopathy (S), and inflammatory/immune processes (I). 13 Since AD and co-pathology most frequently occur in the elderly, V and S biomarkers are crucial for diagnosing and staging AD.
This review will define the AT(N) and NSD-ISS systems as complementary approaches aimed at improving the diagnosis and understanding of LBD. It emphasizes the significant role of LB-AD pathology and neural α-syn disease in clinical phenotypes, disease progression, and overall prognosis of LBDs. Second, this review focuses on recent advancements in fluid biomarkers that can enhance the diagnostic sensitivity for LBD-AD and facilitate rapid screening of patients with co-pathology. Novel fluid biomarkers, such as α-synuclein seed amplification (α-syn SAAs), are discussed for their prognostic, research, and diagnostic utility.
Review criteria
Articles published between 2000 and 2024 were reviewed concerning staging and markers in PD and LBD. Databases such as PubMed and Scopus were searched using the following Boolean search string: ((α-synuclein) OR, neuronal α-synuclein disease integrated staging system NSD-ISS) OR (AT(N) OR (Lewy pathology) OR (Alzheimer’s disease pathology) OR (CSF Aβ42), OR (pTau protein), OR (pTau181), OR(pTau231), (GFAP), OR (YKL-40), OR (fluid biomarkers), OR (Neurogranin), OR (Protein Misfolding Cyclic Amplification (PMCA) OR (Real-Time Quaking-Induced Conversion (RT-QuIC)), OR (tTau), OR (Plasma pTau181), OR (cerebrospinal fluid (CSF) OR (APOE ϵ4alleles), OR (amyloid-/tau-PET)) AND (Parkinson’s disease) AND (Lewy body disorders) AND (Parkinson’s disease with dementia).
No additional filters were applied in the literature search.
Inclusion and exclusion criteria
The inclusion criteria comprised original research papers, reviews, and meta-analyses published in peer-reviewed journals in the English language. The exclusion criteria included:
Books, book chapters, peer-reviewed papers, and publications issued as “letters,” “comments,” “perspectives,” “case reports,” or “surveys.”
Studies of non-English literature.
Research on syndromes other than LBD.
Research that collected data without staging and markers in PD and Lewy body disorders.
Research without human participants.
The snowballing procedure was employed to screen the references of each selected article for potential additional papers that could support the current key evidence, followed by a full-text evaluation of pertinent research. For the narrative synthesis, the structure was based on a discussion of the AT (N) and NSD-ISS staging systems and related fluid biomarkers for LBD diagnosis, followed by an examination of their limitations and potential future directions.
AT (N) system and co-Lewy body – Alzheimer’s disease pathology in Lewy body disorders
The ATN biological staging systems are widely used in research settings to classify the neuropathological features of LBD.12–14 People with PD are categorized as ATN-A (amyloid-positive), ATN-T (tau-positive), or ATN-N (alpha-synuclein-positive) based on the presence of beta-amyloid, tau, and α-syn pathologies. 14 The latest neuropathological consensus criteria for assessing Lewy pathology in post-mortem brains have identified five primary classifications based on their topography: olfactory-pure, amygdala-dominant, brainstem-dominant, limbic/transitional, and neocortical/diffuse Lewy pathology. 15 Lewy pathology often progresses in a caudo-rostral pattern, starting from the lower brainstem and moving to the midbrain/forebrain, with later stages, affecting the neocortex.15,16 This framework accounts for 50%–90% of LBD cases across several studies, primarily reflecting PD patients with an early onset of the disease, a prolonged illness course, and a predominance of motor symptoms.17–20
The ATN has been utilized to evaluate Lewy bodies +AD pathology in DLB and PDD patients, suggesting that DLB patients may either not develop Parkinsonism or may develop it later. Patients with mixed LBD-AD may exhibit specific cognitive deficits, such as memory and language difficulties, which are often less pronounced in cases of pure LBD. 18 The relative contributions of each pathology Lewy-type, Aβ, and tau to the onset of dementia in LBD remain unclear.21–25 Some studies indicate significant involvement of AD co-pathology, particularly the presence of high tau burden.23,25–28 The burden of tau pathology in LBD correlates with global cognitive scores, such as those assessed by the Mini-Mental State Examination and the Boston Naming Test, as well as various neocortical cognitive tasks. 24 Research has shown that the involvement of the Lewy bodies in the limbic system, and especially in the neocortex, plays a key role in cognitive decline in LBD, independent of any neuropathological changes associated with AD.6,14,21,27,29
Factors such as age, gender, APOE and GBA1 genotypes, and the severity of Lewy pathology significantly influence the risk of concurrent AD in LBD.1,9,10 APOE ϵ4 alleles, known risk factors for AD, may be associated with mixed LBD-AD.30–32 The APOE genotypes may modify α-syn aggregation, influencing both AD co-pathology and the course of LBD. Utilizing the ATN framework can assist clinicians in identifying individuals at increased risk and adjusting treatment plans accordingly.
Fluid Alzheimer’s disease biomarkers’ diagnostic and prognostic significance in Lewy body disease
The AT(N) system indicates that CSF concentrations of Aβ-type, phosphorylated tau protein, and total tau protein can be used to support the diagnosis of AD in clinical settings. Notably, AD co-pathology is more prevalent in LBD than in findings from post-mortem investigations.33–36 The percentage of cases with A+T+ CSF as a biological signature of AD is as follows: 2%–10% for PD, 10%–30% for PDD, and 25%–40% for DLB.37–41 A multicenter study conducted on DLB found that the distribution of AT(N) categories, when combined with PET imaging, was approximately 40% for A−T−, 30% for A+T−, 15% for A−T+, and 15% for A+T+. Lower levels of soluble CSF Aβ42 are correlated with both global cognitive deterioration and specific cognitive domain declines, and they may predict the onset of dementia.42–48 While there are no significant correlations between motor impairment and specific clinical subtypes or among different study groups, motor symptoms are associated with more aggressive, or malignant subtypes of PD.42–44,49–53 The concentrations of CSF pTau and tTau are predicted to be slightly altered in AD patients compared to controls, similarly to the Aβ class.38,44,54,55 Higher levels of pTau in CSF are associated with worse outcomes and a reduction in core Parkinsonian symptoms.55–57 However, the association with general motor deterioration, cognitive impairment, and other clinical characteristics is inconsistent.52,53,58–65 Studies have shown that CSF pTau levels are significantly lower in PD cases compared with control cases, but these lower levels may still contribute to cognitive decline. The clinico-pathological significance of these conclusions remains unclear, as later stages of NFTs and Lewy pathologies are associated with reduced levels of CSF Aβ.35,36 Additionally, CSF pTau181 concentrations do not accurately reflect the severity of NFTs, Lewy bodies, and Aβ plaques in AD. In patients with LB pathology and NFTs, CSF levels of t-tau are consistently elevated.33,34,36 CSF AD biomarkers in LBD detect patients with co-pathologies and advanced stages of Lewy pathology, with increased risk of cognitive deterioration. Blood pTau levels are strongly correlated with neuropathologic alterations, pathological CSF biomarker levels, and PET positivity in AD. Studies examining blood biomarkers in LBD have often found that changes in blood levels of Aβ and tau proteins are minimal or absent.66–85 (Table 1) Skin pTau181 and pTau231 levels were slightly higher in PD and DLB patients compared to controls.79,85–87 However, both pTau231 and pTau-PET, as well as skin and CSF levels were found to be lower in these patients than in control cases.75,85 Plasma pTau181 can differentiate DLB and PDD patients from without CSF abnormalities. Future research should provide more insight into the pathological causes behind blood pTau concentrations in LBD and their potential benefits for recruitment in clinical trials. Novel fluid biomarkers have been studied in LBD patients to better evaluate the presence of AD co-pathology in vivo. These include measures of α-syn concentrations, neuroaxonal damage markers such as neurofilament light chain protein (NfL), synaptic dysfunction markers like neurogranin, β-synuclein, and synaptosomal-pathological protein25 (SNAP-25), and indicators of neuroinflammation such as glial fibrillary acidic protein (GFAP) and chitinase-3-like protein 1 (YKL-40).76,83,84,88–90 (Figure 1) Both α-synuclein real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA) are extremely sensitive methods for detecting αSynD in various biospecimens, including brain tissue, CSF, skin, and olfactory mucosa from LBD patients. 91 CSF α-syn RT-QuIC has been found to be highly comparable to results from post-mortem biopsies of LBD patients, even in cases where AD co-pathology is present.92,93 Large-scale studies, both in those with and without cognitive decline, indicated that CSF α-syn RT-QuIC positivity was observed in 23% and 8% of cases, respectively, with Lewy and AD co-pathology detected in nearly 11% and 2% of cases, respectively.94–100 However, more research is needed to enhance inter-laboratory reproducibility and consistency of protocols for identifying SAAs in clinical practice. Several studies have reported lower levels of total CSF α-syn in LBD compared to other neurodegenerative diseases and controls. To improve diagnostic accuracy, specific pathological forms of α-syn, such as phosphorylated α-syn, have been examined. The effectiveness of certain measurements of α-syn for direct clinical application in PDD/DLB and AD is inconsistent.101–109 The long-term progression in PD may suggest that higher levels of oligomeric CSF α-syn are more closely associated with non-motor symptoms. Attempts to detect peripheral tissue synucleinopathy via skin biopsies and saliva have shown slightly poorer accuracy than CSF RT-QuIC protocols.107–109 Additionally, blood-based measurements of oligomeric α-syn concentrations are elevated by quantifying α-syn and SAAs in blood samples.110–114 NfL, a key component of the axonal cytoskeleton, is widely recognized as a fluid biomarker of neuro-axonal disorders.115–117 In DLB and its prodromal stage, elevated levels of NfL in the blood are associated with worse motor and cognitive outcomes, and they can predict the transition from prodromal to PD and full-blown DLB. However, the range at which AD co-pathology affects CSF and blood levels of NfL remains uncertain. Large studies involving neuropathological or biomarker-enhanced samples are needed to clarify the role of NfL in LBD-AD.118–122 Potential fluid biomarkers GFAP and YKL-40 can both indicate neuroinflammation/endothelial dysfunction and astrocytic stimulation. The CSF and blood samples from individuals with AD showed higher levels of GFAP, indicating that astrocytic activation is being accelerated alongside Aβ processes.123–127 Plasma GFAP concentrations were found to be higher in advanced DLB compared to earlier stages, and they were significantly elevated in cases with positive amyloid and tau PET scans compared to those with negative scans.84,85 Preliminary findings regarding new biomarkers of synaptic dysfunction and disruption were validated, showing that levels of neurogranin, SNAP, and synuclein proteins in AD and LBD-AD groups compared to the control and LBD groups without AD pathology.128–133 However, these findings have not yet been replicated in blood due to technical issues with immunoassays.
Overview of main studies of core Alzheimer’s disease biomarkers in blood samples of patients with Lewy body disease.

Aβ, amyloid-β; AD, Alzheimer’s disease; ADNC, AD neuropathological changes; DLB, dementia with Lewy bodies; IRBD, isolated rapid eye movement (REM) sleep behavior disorder, LBD, Lewy body disease; MCI, mild cognitive impairment; PD, Parkinson’s disease; PDD, Parkinson’s disease with dementia; pTau, phosphorylated tau; Simoa, single molecule array; tTau, total tau.
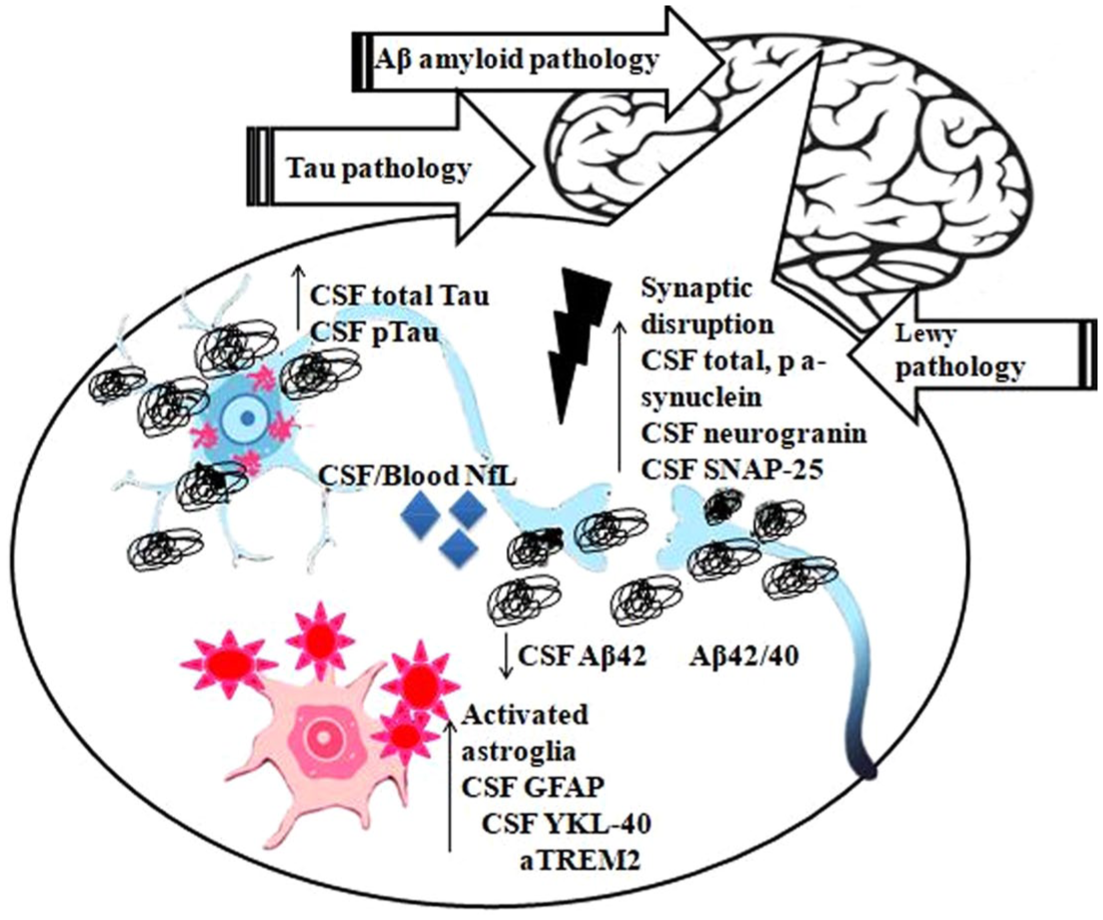
Fluid biomarkers and main pathophysiological mechanisms of Lewy body disease with Alzheimer’s disease co-pathology.
Biological definition of neuronal αsyn: The neuronal α-synuclein integrated staging system
Simuni et al. 5 introduced the term “neuronal synuclein disease” (NSD) as part of their Neuronal Synuclein Disease Integrated Staging System (NSD-ISS), taking an unconventional approach to understanding this condition. The defining characteristic of NSD is the presence of aggregated synuclein (S) and the potential for neuronal dysfunction (D, i.e., degeneration) within the NSD-ISS. SAA conducted on CSF and molecular DAT imaging are reliable methods for assessing the status of S and D, respectively. A key component of this framework is the complete overlap of disease phenotypes.
The NSD-ISS does not differentiate between individuals who exhibit early cortical contribution (as seen in DLB) and those who do not (as in PD). Furthermore, if there is insufficient evidence of S, individuals who meet the criteria for a PD diagnosis are not eligible for an NSD diagnosis. This limitation means that the NSD-ISS does not accommodate the many individuals who have with genetic mutations such as PRKN or LRRK2. On the other hand, asymptomatic individuals misclassified as S+ may actually suffering from NSD. Patients classified as S+ in stage 1 of NSD are asymptomatic, while those in stages 2 and 3 exhibit various levels of clinical severity including motor and mild/non-motor. The authors noted that this staging should incorporate other biological features that have yet to be fully identified.
Alternately, Höglinger et al. 4 discussed the synergistic effects of synuclein (S), neurodegeneration (N), and genetic (G) factors in their proposed classification system, referred to as SynNeurGeon. They emphasized that validated tests, such as skin biopsies or CSF analyses, should serve as the basis for determining the S+ or S-component in the biological categorization. Molecular imaging can reveal pathological patterns in dopaminergic, cortical, or and even cardiac/sympathetic associated with PD. Importantly, motor symptoms are not required for a biological definition of the disease. The authors clearly differentiated between sporadic PD (S+, N+) and sporadic Parkinson’s type synucleinopathy (S+, N−), highlighting the importance of understanding the S+/N status, which remains unclear.
Although it is not currently understood how these individuals progress to clinical disease, the proposed biological classification acknowledges the presence of asymptomatic S+ people with Parkinson’s type (or Lewy-type) synucleinopathy.
The authors extended the genetic classification to include predisposing genetic factors for PD, like GBA1 and LRRK2, alongside fully penetrant variants such as SNCA and PRKN.
AT(N) and NSD-ISS systems as complementary approaches to diagnose LBD
The NSD-ISS framework and the AT(N) system are two research tools used to better categorize and comprehend LBD. While the AT(N) system, which is more widely used in Alzheimer’s research, classifies pathology based on amyloid, tau, and neurodegeneration biomarkers, the NSD-ISS concentrates on staging based on the presence and progression of neuronal α-syn pathology. 135 These systems can potentially complement each other. By utilizing both frameworks, clinicians may be able to identify LBD at earlier stages, even before the onset of significant clinical symptoms. 136 The AT(N) system can be used to assess the extent of amyloid and tau pathology in individuals with LBD who are also being staged using the NSD-ISS framework. 137 Optimizing the categorization and selection of patients with LBD for clinical trials is the goal of both the NSD-ISS and AT(N) systems.138,139 by incorporating biological markers, as already has been discussed for AD fluid biomarkers in the previous section. Next, novel α-syn SAAs will be evaluated as potential fluid biomarkers for LBD.
α-synSAAs as novel fluid biomarkers of neuronal α-syn
Any rigorously verified n-α syn biomarker can identify neuronal α-syn disease. 5 For over 20 years, in vivo measurements and detection of pathogenic α-syn have been conducted with promising results. 140 However, the only test that has been fully validated to date is the CSF α-syn SAAs test. Table 2 summarizes studies on α-syn SAAs as novel fluid biomarkers of neuronal α-syn.
Overview of studies on α-syn SAAs as novel fluid biomarkers of LBDs.
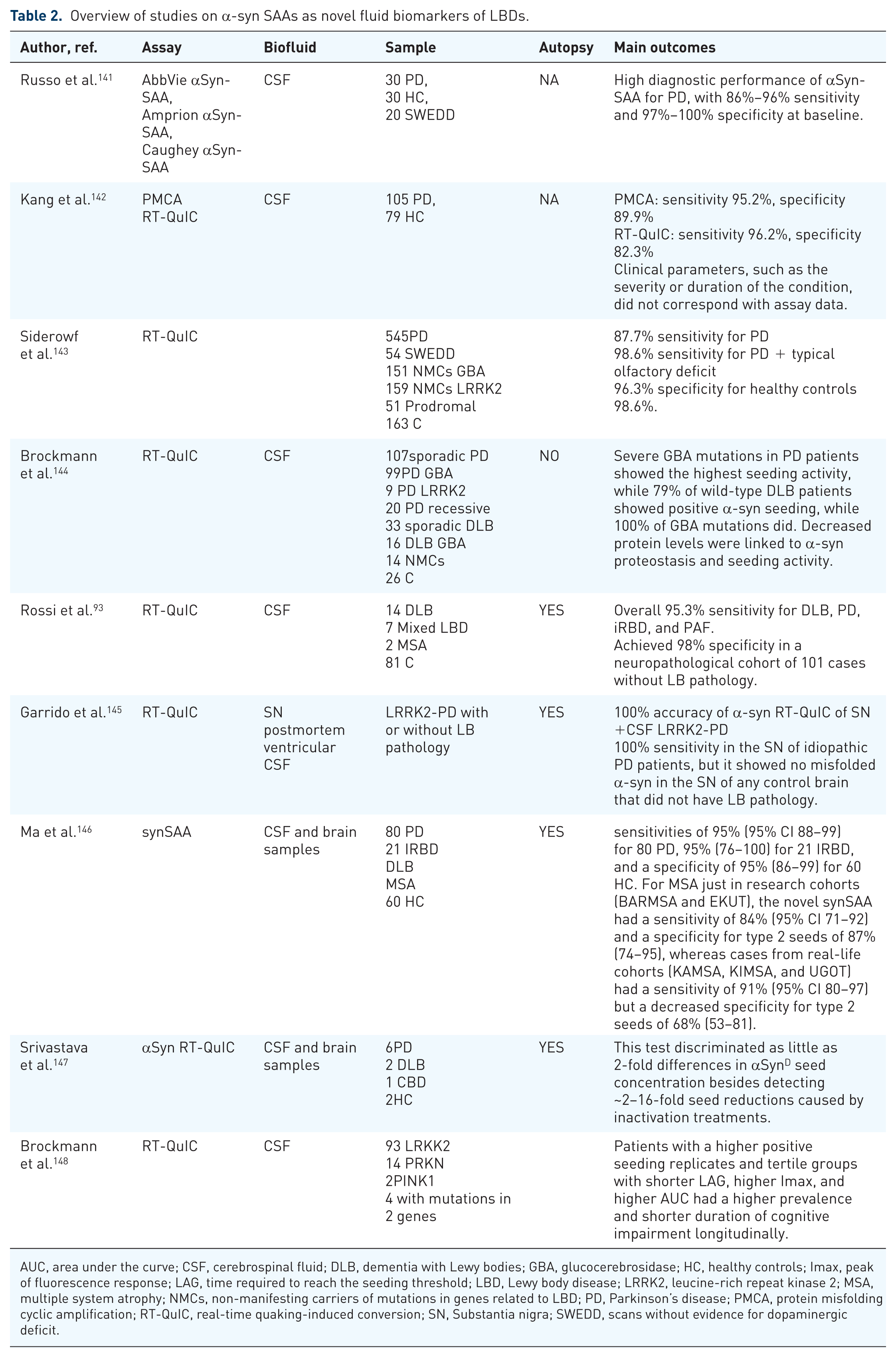
AUC, area under the curve; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; GBA, glucocerebrosidase; HC, healthy controls; Imax, peak of fluorescence response; LAG, time required to reach the seeding threshold; LBD, Lewy body disease; LRRK2, leucine-rich repeat kinase 2; MSA, multiple system atrophy; NMCs, non-manifesting carriers of mutations in genes related to LBD; PD, Parkinson’s disease; PMCA, protein misfolding cyclic amplification; RT-QuIC, real-time quaking-induced conversion; SN, Substantia nigra; SWEDD, scans without evidence for dopaminergic deficit.
The CSF α-syn SAAs were explored as a possible alternative to conventional CSF analysis. Originally developed for the detection of PrPsc in prion disorders, “Protein Misfolding Cyclic Amplification (PMCA)” and “Real-Time Quaking-Induced Conversion (RT-QuIC)” are two ultrasensitive protein amplification methods used to detect pathological protein aggregates. 149 These techniques mimic protein aggregation and misfolding in CJD, similar to the polymerase chain reaction (PCR), where the number of template units increases while the quantity of the substrate decreases. 149 Although both methods follow the same principle, they differ significantly in key aspects. In RT-QuIC, a vigorous, intermittent shaking action is used to induce protein aggregation, while PMCA relies on sonication for the same purpose. At the end of each procedure, PMCA utilizes Western blot analysis and PK digestion to detect and characterize abnormal aggregates. The formation of these aggregates is monitored in real time by their ability to bind a fluorescent dye, called thioflavin T (ThT). The major strengths of this biomarker are: (i) the ability to distinguish different synucleinopathies, as the assay can distinguish the neuronal α-syn pathology (such as PD and DLB) and the pathology predominantly involving glial inclusions in multiple system atrophy (MSA); (ii) the potential to differentiate between synucleinopathies and tauopathies, although further optimization is recommended; and (iii) the potential for quantitative assay use that could provide prognostic data by accounting for SAA kinetics.
The assay has been validated by consistent outcomes in two different studies using well-characterized samples across multiple independent laboratories.141,150 Therefore, CSF α-syn SAA provides the level of evidence required for a precise diagnosis of α-syn disease. This test reliably detects patients with clinical presentations of PD and DLB, showing a positive result in over 95% of autopsied cases that have been confirmed.91,142–144,151 The SAA can identify individuals at risk for clinically recognized conditions, such as idiopathic rapid eye movement sleep behavior disorder, olfactory dysfunction, and inherited risk factors. Positive CSFα-syn SAA results have been linked to clinically diagnosed α-synucleinopathy within 4–6 years, both in patients with isolated hyposmia and those without cognitive impairment.
Several studies have shown that both PMCA and RT-QuIC assays can identify patients with LBD with high sensitivity and specificity.93,94,151 RT-QuIC tests showed a sensitivity of 100% for BH samples and 83% for CSF samples. 145 A meta-analysis indicated that PMCA may be more effective than RT-QuIC for detecting MSA aggregates. 146 However, technical optimization or staff performance issues may affect the results.
The novel Amprion’s SAAmplify-αSYN Biomarker Test represents a significant breakthrough because it reliably distinguishes MSA from other synucleinopathies. 146 The study classified synuclein seeds based on their fluorescence levels: seed type 1, which is associated with PD, DLB, and idiopathic REM sleep behavior disorder (iRBD), exhibits high fluorescentity. In contrast, seed type 2, associated with MSA, shows a moderate fluorescence. Conducted across seven institutions in four countries, the study achieved 100% compliance with the gold standard of pathology for brain and CSF samples. The novel syn SAA confirmed a precise diagnosis based on biomarker by achieving 95% sensitivity for PD and IRBD and 95% specificity for healthy controls. 146 Type 1 α-syn seeds were found in all samples from patients with PTSD and with a history of severe dementia. These results highlight syn SAA’s potential for high-accuracy patient stratification, differential diagnosis, and early detection of MSA.
Limitations
One of the disadvantages of the ATN biological staging method is the inconsistent evaluation of neuropathological characteristics. Several studies may classify people based on aberrant brain proteins using different criteria and techniques, which could lead to conflicting results.12,13 To guarantee the accuracy and repeatability of results, neuropathological assessment and classification criteria must be standardized.
The ATN system’s reliance on post-mortem brain imaging to evaluate the co-pathology of the Lewy bodies presents a significant drawback. While postmortem investigations provide valuable insights into the neuropathological characteristics of neurodegenerative diseases, they are impractical for daily clinical application. One promising avenue of research circumvent is the development of non-invasive imaging methods and biomarkers to assess Lewy body co-pathology in living subjects.
Additionally, current biomarkers for AD have limitations, including the lack of approved biofluid reference supplies and techniques, with the exception of CSF Aβ42 and Aβ40. When diagnosing early or mild ADNPC, biomarkers, such as blood, CSF, and PET, show lower sensitivity compared to neuropathological examinations. 12 Furthermore, neuropathological staging and disease assessment through PET (or liquid biomarkers) are not congruent. For example, tau PET ligand uptake varies across different Braak stages, which does not align with the Braak neuropathological staging. The limitations of these biomarkers may be viewed as both a weakness and a virtue, as aberrant Core 1 biomarkers suggest that ADNPC is more likely to be present in general, rather than only in neuritic plaques. Currently, there is a lack of well-studied biomarkers for various diseases, making it impossible to determine in vivo the presence of other illnesses or to assess their specific impacts adequately.12,13 Consequently, it is not possible to determine the exact proportion of cognitive decline in a person that is related to AD compared to other neuropathologic conditions. Clinical evaluations and biomarker results can provide probabilistic estimates.
However, SAAs still have a number of limitations. SAAs currently provide contradicting data with different cut-offs, despite the fact that several studies have shown the significance of quantitative indicators.144–146,152 Another issue with SAAs is their specificity in detecting α-syn pathology, which may restrict their applicability in diagnosing other neurodegenerative diseases that have similar protein aggregation patterns. One of the difficulties in using α-syn as a biomarker to distinguish PD from other neurodegenerative diseases is the variability in the causes of PD. Additionally, research has demonstrated that total α-syn levels in CSF often lack sufficient sensitivity and specificity, making it difficult to differentiate between PD, DLB, and AD.90,153 Furthermore, the lack of standardization in α-syn serum protocols has also presented difficulties. 152 Another challenge is developing study protocols that more accurately detect protein aggregation. Current methods for detecting α-synare limited by factors such as low sensitivity, high costs, and the need for complex sample preparation. Fluorescence microscopy provides high sensitivity and specificity, enabling the visualization of fluorescent constituents even in small quantities. 154 Additionally, emerging imaging techniques like mass spectrometry and super-resolution microscopy can provide precise information on the structure of protein aggregation. 93 End-point dilution is used to measure seed concentration, but there have only been a few studies conducted on sequential dilutions.147,149 Factors such as detergents and blood contamination can affect the kinetic parameters and the number of positive replicates in RT-QuIC assays. The reproducibility of results improves with a higher number of replicates. Among the kinetic measure, the time to threshold (LAG) is the most reliable. 148 Future research should focus on developing selective SAAs to detect a broader range of pathological protein aggregates.
Future perspectives
Neurodegenerative research is increasingly using biological approaches to diagnose and define LBDs rather than just relying solely on clinical presentations. The emphasis on biologically based diagnosis and staging is transitioning from being exclusive to research to becoming essential for both clinical treatment and research purposes.
To establish relevant associations at the community level, it is crucial to conduct real epidemiological and collect real-world data on biomarker properties in representative populations.
Standardization of cut-off points, tau PET measurement techniques, and biofluid tests is necessary. As with other disorders, the precise threshold value for abnormalities may change over time as more data becomes available to assess the prognosis.
A better comprehension of the various post-translational changes of tau could enhance fluid-based biological staging.
Biomarkers will play a vital role in the bio-characterization and prediction of AD pathophysiology, especially if unique blood abnormalities can be detected.
In clinical trials that focus on pathways other than anti-Aβ immunotherapy, future criteria should evaluate how therapies influence biomarkers and clinical outcomes.
Despite being somewhat unrealistic at present, the development of multiplex blood-based marker tests could eventually facilitate a comprehensive AT1T2NISV diagnostic and staging system, possibly including TDP-43.
Regional variations exist in the applicability of these criteria to diagnose and screen for AD in clinical practice, even among high-income nations listed by the World Bank.
In low- and middle-income nations, blood-based biomarkers for AD diagnosis are expected to become increasingly relevant as they become more affordable and accessible, potentially surpassing the use of PET and CSF-based biomarkers. This shift is driven by the high cost and limited availability of PET and CSF-based tests in these regions.
Conclusion
The ATN biological staging system assists in identifying patients at risk of disease progression and tailoring treatment plans. Future research should aim to standardize neuropathological assessments and advance non-invasive biomarkers.