
Abstract
Orthotopic liver transplantation (OLT) was the first treatment able to modify the natural course of hereditary transthyretin (ATTRv) amyloidosis, which is a rare and fatal disorder caused by the accumulation of misfolded transthyretin (TTR) variants in different organs and tissues and which leads to a progressive and multisystem dysfunction. Because the liver is the main source of TTR, OLT dramatically reduces the production of the pathogenic TTR variant, which should prevent amyloid formation and halt disease progression. However, amyloidosis progression may occur after OLT due to wild-type TTR deposition, especially in the nerves and heart. In this review, we discuss the disease features influencing OLT outcomes and the clinical manifestations of ATTRv amyloidosis progression post-OLT to improve our understanding of disease worsening after OLT and optimize the follow-up and clinical management of these patients. By conducting a literature review on the PubMed database, we identified patient characteristics that have been associated with worse post-OLT outcomes, including late-onset V50M and non-V50M variants, age >40 years, long disease duration, advanced neuropathy and autonomic dysfunction, and malnutrition. Regarding post-OLT mortality, deaths occurring within the first year after OLT were mainly associated with fatal graft complications and infectious diseases, whereas cardiovascular-related deaths usually occurred later. Considering the diverse clinical manifestations of ATTRv amyloidosis progression post-OLT, including worsening neuropathy and/or cardiomyopathy, autonomic dysfunction, and oculoleptomeningeal involvement, we present advice on the most relevant tests for assessing disease progression post-OLT. Finally, we discuss the use of new therapies based on TTR stabilizers and TTR mRNA silencers for the treatment of ATTRv amyloidosis patients post-OLT.
Introduction
Hereditary amyloid transthyretin (ATTRv) amyloidosis is an inherited, progressive, and life-threatening disease caused by mutations in the transthyretin (TTR) gene. The resulting misfolded TTR proteins form aggregates that are deposited in different organs and tissues, causing their progressive dysfunction. 1 If left untreated, the average duration of survival from disease onset is approximately 10 years.1–4
More than 120 TTR pathogenic variants have been reported to cause ATTRv amyloidosis.4,5 Most are associated with a neurological phenotype or a mixed phenotype with neurological and cardiac involvement, although certain TTR variants can present with a predominantly cardiac phenotype.4,5 The predominant genotype, Val50Met (V50M), is responsible for ATTRv amyloidosis in endemic foci, 1 where it is typically associated with early-onset disease (age < 50 years), small-fiber neuropathy, and frequent autonomic symptoms, including sweating disorders, gastrointestinal and urinary tract disorders, orthostatic hypotension, and sexual dysfunction. 6 In contrast, in non-endemic areas, patients with the V50M variant usually have late-onset disease (age ⩾ 50 years) with more severe motor neuropathy and milder autonomic dysfunction than early-onset V50M patients.1,4,6 Along with late-onset V50M, some non-V50M variants, such as Ile127Val, Thr80Ala, and Ser97Tyr, have been associated with increased disease severity and lower life expectancy. 7
The first treatment for ATTRv amyloidosis was orthotopic liver transplantation (OLT), which was considered the gold standard since it was introduced in the 1990s until the approval of pharmacological agents capable of modifying disease progression. Because 95% of TTR protein is produced in the liver, OLT aims to prevent amyloid formation and halt disease progression by replacing the main source of pathogenic TTR with an organ that produces wild-type TTR. 8 Globally, more than 2000 ATTRv amyloidosis patients have undergone OLT, 9 with approximately half of them from Portugal, the country with the largest endemic cohort of early-onset V50M patients. 1 Although the procedure improves survival in patients with ATTRv amyloidosis,9–11 the clinical benefit depends on patient characteristics, and OLT sometimes fails to stop subsequent disease progression due to the continued deposition of wild-type TTR in the nerves and heart.1,9,11 Consequently, a better understanding of disease progression after OLT is crucial to identify the particular needs of these patients and to facilitate the development of a specific strategy to monitor them and improve their clinical management.
In this publication, we review the disease features influencing OLT outcomes and the clinical manifestations of disease progression post-OLT and present advice on the follow-up and management of ATTRv amyloidosis patients who undergo OLT based on the literature and our experience with these patients.
Methods
In November 2021, key opinion leaders on ATTRv amyloidosis met to discuss the current strategies for evaluating disease progression in ATTRv amyloidosis patients who undergo OLT.
A literature review was performed using the PubMed database with no time limit and with the following search terms: hereditary transthyretin amyloidosis, ATTRv, hATTR, transthyretin familial amyloid polyneuropathy, transthyretin amyloid neuropathy, liver transplant, or treatment. The main objective of the literature search was to assess outcome data of liver transplantation in ATTRv amyloidosis patients and to identify the reported clinical manifestations of disease progression after surgery. To this end, the data were obtained from reviews and original research articles related to these topics; clinical case reports were not included in the analysis.
Based on the published data and the clinical experience of the key opinion leaders, this publication aims to guide the follow-up and management of patients with ATTRv amyloidosis after OLT.
Results
Profile of patients with ATTRv amyloidosis undergoing liver transplantation
Several reports concerning the effects of OLT on ATTRv amyloidosis describe a worse outcome of this procedure in patients with late-onset V50M and non-V50M variants.3,10–13 Thus, patients with the early-onset V50M variant are the most common candidates for OLT. Ideally, OLT should be performed at early disease stages to provide a greater clinical benefit.3,9 According to the published data, disease duration from onset to OLT ranges from 2 to 5 years on average.3,10–13 Because OLT does not usually reverse the existing neuropathy and organ impairment,1,9 an early intervention is required to avoid the development of severe symptoms and provide patients with an acceptable quality of life. In this regard, the main challenges are diagnostic delays due to the multisystemic and nonspecific clinical manifestations of the disease, its rarity, and prolonged waiting times until OLT. With the advent of novel therapies based on TTR stabilizers and TTR mRNA silencers, another relevant challenge in the near future may be the risk of delaying OLT in patients with disease progression despite specific treatment.
Peri-transplant and long-term morbidity and mortality in patients with ATTRv amyloidosis post-OLT
Patients’ status at the time of OLT largely influences their survival. In particular, malnutrition and a longer disease duration before OLT have been associated with elevated mortality. 14 Importantly, previous studies reported that mortality risk increases by 11−17% per year of disease progression before OLT,3,11 which reinforces the need for early intervention.
Although some studies failed to establish a correlation between neurological dysfunction and post-OLT survival,14,15 severe sensorimotor neuropathy and autonomic dysfunction have been associated with elevated post-OLT mortality. 12 In addition, late-onset disease has been associated with progressive cardiomyopathy and higher post-OLT mortality, especially in males.9,16
Perioperative complications within the 90-day post-OLT period include bleeding, bile leak, bile duct strictures, renal dysfunction, thrombosis (including hepatic artery thrombosis), graft failure, arrhythmia, and infections. 17
Deaths occurring within the first year post-OLT have been mainly associated with fatal graft complications, which include hepatic failure, hemorrhagic shock, and mesenteric ischemia, and infectious diseases, including opportunistic infections. 18 In contrast, deaths related to cardiovascular diseases, which are the leading cause of mortality post-OLT and mainly comprise heart failure and sudden cardiac death (probably due to continued deposition of wild-type TTR in the heart), and strokes (which are probably caused by the deposition of variant TTR produced in the choroid plexus), occur later than those associated with graft rejection or infection 18 (Table 1). These results are consistent with the clinical outcomes of 42 patients who underwent OLT at Bellvitge University Hospital, Barcelona, Spain, from January 1997 to August 2019 (Table 2).
Main causes of early and late deaths in ATTRv amyloidosis patients after liver transplantation. 18
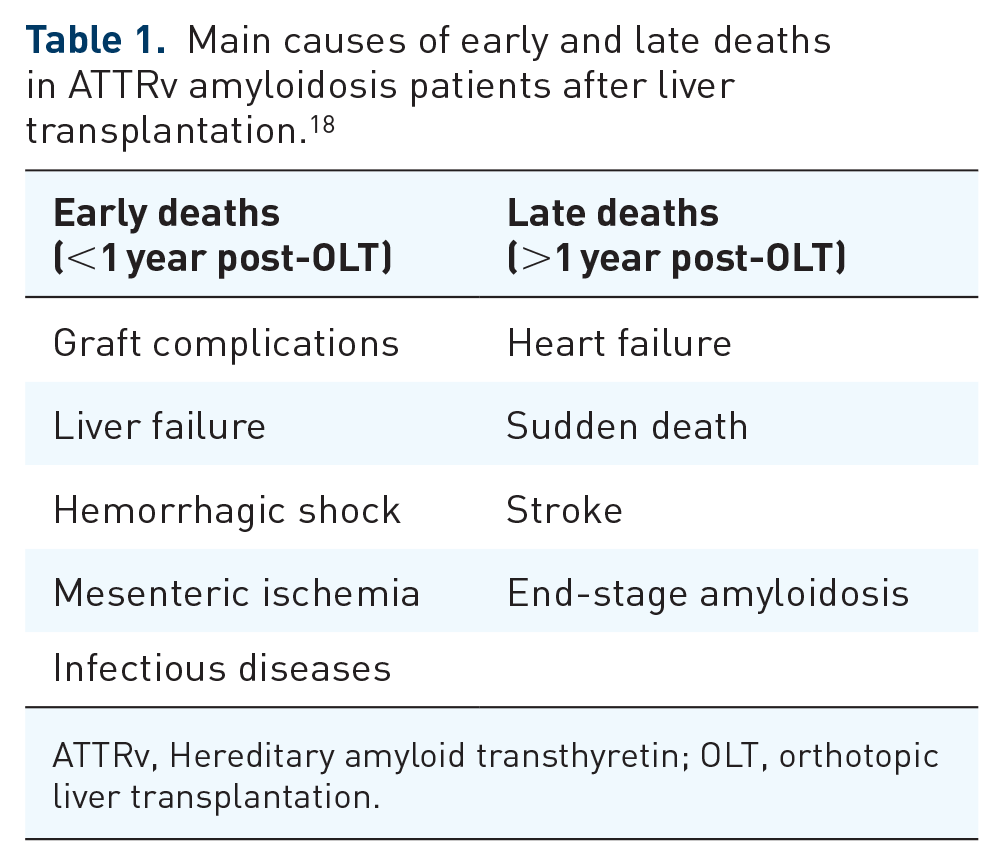
ATTRv, Hereditary amyloid transthyretin; OLT, orthotopic liver transplantation.
Characteristics and clinical outcomes of patients who underwent OLT at Bellvitge University Hospital from January 1997 to August 2019.

Values are mean ± standard deviation and (median) or number of observations and (%).
ATTRv, hereditary amyloid transthyretin.
Factors associated with better OLT outcomes in ATTRv amyloidosis patients
Previous analyses of the long-term outcomes of ATTRv amyloidosis patients proved that OLT has a major survival benefit.3,9,20 Indeed, more than 50% of ATTRv amyloidosis patients have a 20-year post-OLT survival rate, 11 which is a significant improvement compared with the natural course of the disease. However, OLT outcomes highly depend on the TTR variant and disease characteristics. For instance, the 5-year post-OLT survival rate has been reported to be approximately 100% for V50M patients and 59% for non-ATTR V50M patients. 9 In addition, the clinical outcome of OLT was found to be especially favorable in early-onset V50M patients compared with late-onset V50M patients10,11 (Table 3).
Factors influencing the clinical outcome of liver transplantation in ATTRv amyloidosis patients.

mBMI, modified body mass index.
Timely interventions at early disease stages, that is, in the absence of pronounced symptoms of neuropathy, autonomic dysfunction, and/or cardiomyopathy, are associated with better prognosis and are the main recommendation to ensure patients’ quality of life.12,14 In addition, better OLT outcomes are observed when patients undergo surgery within the first 7 years of disease onset and still have good nutritional status, with a modified body mass index (mBMI) ⩾600 kg/m2·g/L9,14,21 (Table 3).
Finally, age is another factor to be considered before OLT. In this regard, patients who undergo surgery before 40 years of age show better outcomes than those who undergo the OLT later 21 (Table 3).
Progression of ATTRv amyloidosis post-OLT
Although OLT results in rapid clearance of the pathogenic TTR variant from circulation, there are several reports of disease progression due to the incorporation of wild-type TTR into preexisting amyloid deposits.1,9,22,23 Indeed, worsening cardiomyopathy and progressive sensorimotor polyneuropathy and cachexia are the main causes of death after the first post-OLT year. 18
Continued wild-type TTR deposition after OLT can affect different organs and tissues, including the kidneys and nerves,24,25 but it preferentially occurs in the heart. 24 Because cardiac amyloid is more prone to wild-type TTR accumulation, progressive post-OLT cardiomyopathy mostly affects patients with previous cardiac involvement, that is, patients with late-onset V50M and non-V50M variants associated with preexisting cardiac TTR deposits.10,23 However, it can also affect patients with early-onset V50M. 26 Interestingly, the amyloid fibrils of these patients comprise a mixture of full-length and truncated TTR (type A), whereas only full-length TTR (type B) is found in the fibrils of early-onset V50M patients with a predominantly neurological phenotype.10,22,27 Wild-type TTR is more easily incorporated into type A fibrils than into type B fibrils, which explains the increased susceptibility of type A patients to post-OLT progressive cardiomyopathy.9,10,23,28
Symptoms of disease progression following OLT
Because the extrahepatic sources of TTR – the retina and choroid plexus – hardly contribute to the circulating TTR pool, abnormal protein levels are dramatically reduced in the blood after OLT. 29 As a result, some patients experience improved survival and quality of life but, unfortunately, other patients have worse outcomes.12,29 Although OLT is of less benefit in patients with non-V50M variants, neuropathy and/or cardiomyopathy progression can occur even in V50M patients.26,29,30
Progressive cardiac amyloidosis after OLT is a major concern because the continued deposition of wild-type TTR can develop as restrictive cardiomyopathy in both non-V50M and V50M patients.24,26,29,31 In addition, a substantial percentage of post-OLT ATTRv amyloidosis patients experience a worsening of peripheral and autonomic nerve symptoms, with the progression of the sensorimotor deficit and autonomic neuropathy, including gastrointestinal symptoms, bladder dysfunction, and life-threatening arrhythmias.12,25,29 Moreover, as in other patients who have undergone liver transplantation, renal function often deteriorates in ATTRv amyloidosis patients after OLT. 32 However, a beneficial effect of OLT on kidney amyloidosis has been observed in some patients 32 and, although continued wild-type TTR deposition in the glomeruli may cause kidney damage, 24 the toxic effects of the immunosuppressive treatment may also contribute to renal dysfunction. 32
Ocular and cerebral manifestations due to oculoleptomeningeal amyloidosis are other important aspects of disease progression post-OLT. Because OLT has no impact on the production of variant TTR in the eye and choroid plexus, ocular and leptomeningeal amyloidosis continues due to the deposition of locally produced variant TTR.29,33 Although tonic–clonic seizures and transient focal neurological episodes have been related to cerebral amyloid angiopathy in post-OLT patients,34,35 severe ocular manifestations include vitreous opacities and glaucoma. 36 Importantly, the frequency of the latter has increased due to the improved survival of post-OLT patients, and it affects over 40% of patients who have undergone OLT after a disease duration of five or more years. 36
Use of specific drugs for ATTRv amyloidosis in post-OLT patients
Currently, TTR stabilizers and TTR mRNA silencers are approved for the treatment of ATTRv amyloidosis. 37 Several case reports on the use of the TTR stabilizer tafamidis in post-OLT patients with neurological deterioration showed diverse outcomes that depended on the TTR variant but no safety issues.19,38,39 In a post-OLT V50M patient with progressive sensorimotor polyneuropathy and oculoleptomeningeal manifestations, the drug was able to halt clinical deterioration during an 18-month follow-up period. 38 However, a post-OLT patient with the Glu74Gly variant and another with the Leu78Arg variant experienced a continued worsening of sensorimotor neuropathy despite tafamidis treatment, although the drug stabilized the symptoms of autonomic dysfunction in the latter.19,39 Interestingly, the Glu74Gly patient improved after switching from tafamidis to patisiran, 19 a small interfering RNA that targets TTR mRNA transcripts and prevents TTR synthesis in the liver. 40 The positive effects of patisiran in post-OLT patients were further confirmed by a phase IIIb trial that included ATTRv amyloidosis patients with disease progression post-OLT. The 12-month safety and efficacy results of this study demonstrated that patisiran improved neuropathy, quality of life, autonomic dysfunction, and nutritional status from baseline and was well tolerated. 41 Although long-term follow-up data are still needed, the results of this study, which is the first clinical trial of a pharmaceutical agent for ATTRv amyloidosis in post-OLT patients, are consistent with those observed in the clinical trials of patisiran in non-transplant ATTRv amyloidosis patients40,42–44 and therefore support the ability of patisiran to also halt the progression of ATTRv amyloidosis symptoms in post-OLT patients. Notably, patisiran was not associated with hepatotoxicity or allograft rejection and was well tolerated despite the use of immunosuppressants. 41 Another study describing the experience with inotersen, an antisense oligonucleotide that binds to liver TTR mRNAs and represses translation, 45 reported some benefits of the drug in stabilizing neuropathic symptoms post-OLT, but a high rate of adverse events. 46
Discussion
Because disease progression can occur after OLT regardless of the TTR variant, post-OLT ATTRv amyloidosis patients should be closely monitored. The following are our recommendations for the optimal follow-up and clinical management of these patients, considering the diverse outcomes of OLT reviewed above and their special requirements.
Currently, with the available pharmacological treatments for ATTR amyloidosis, OLT has been relegated to a second-line treatment. In many countries where all treatments (stabilizers and gene silencers) are available, OLT is now rarely used. However, some factors, such as patient preferences, the need for another organ transplant (i.e. heart), local pharmacological access, and, more uncommonly, failure of all previous first-line treatments, keep OLT a treatment option for ATTRv amyloidosis.
Prior to OLT, the baseline for the follow-up assessments should be established to have a reference value for successive post-OLT evaluations. These evaluations should include the same tests and investigations as those used to monitor disease progression in non-transplanted ATTRv amyloidosis patients, along with functional evaluation of the transplanted liver by the liver transplant unit.
Periodic follow-up assessments should be performed by a multidisciplinary team to cover the wide range of manifestations of disease progression due to the continued deposition of wild-type TTR after OLT. Special attention should be given to progressive cardiomyopathy after OLT because cardiac events, primarily heart failure, and sudden cardiac death have been reported to be the leading causes of mortality in post-OLT ATTRv amyloidosis patients. 18
The criteria to define disease progression in post-OLT patients should be comparable to those applied to non-transplanted patients. In this regard, the levels of changes in the clinical indicators proposed by Conceição et al. to identify significant disease progression 47 could be used to determine the need for an additional therapeutic intervention in post-OLT patients (Table 4).
Criteria for defining normal and potential changes in the disease course in diagnosed and treated patients with ATTRv amyloidosis, as proposed by Conceição et al. 47

6MWT, 6-minute walk test; ADLs, activities of daily living; BUN, blood urea nitrogen; CMAP, compound muscle action potential; ECG, electrocardiography; ECHO, echocardiography; ESC, electrochemical skin conductance; FAP-RODS, Familial Amyloid Polyneuropathy Specific Rasch-Built Overall Disability Scale; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy; NIS, Neuropathy Impairment Score; NT-proBNP, N-terminal pro-brain natriuretic peptide; PSMS, Physical Self-Maintenance Scale; PND, Polyneuropathy Disability; SNAP, sensory nerve action potential.
A proposed algorithm for the follow-up of ATTRv amyloidosis post-OLT is depicted in Table 5. A general assessment should be performed every 6 months and evaluate orthostatic hypotension, mBMI, polyneuropathy disability (PND) score, and Coutinho stage, along with laboratory tests for assessing liver function, immunosuppression status, and circulating levels of cardiac biomarkers, including N-terminal pro-B-type natriuretic peptide (NT-proBNP) and troponin. Because kidney function often declines after liver transplantation, laboratory tests should also analyze the serum creatinine levels, glomerular filtration rate, and urinary microalbumin to evaluate renal function.
Proposed follow-up strategy for ATTRv amyloidosis patients who have undergone liver transplantation.

Specific post-OLT assessments.
A different small-fiber test can be performed depending on center experience and/or test availability.
COMPASS-31, Composite Autonomic Symptom Score 31; ECG, electrocardiography; ECHO, echocardiography; EMG, electromyography; GFR, glomerular filtration rate; mBMI, modified body mass index; MRI, magnetic resonance imaging; NT-proBNP, N-terminal pro-brain natriuretic peptide; NIS, Neuropathy Impairment Score; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy; SFN/SIQ, Small-Fiber Neuropathy-Symptoms Inventory Questionnaire; PND, polyneuropathy disability; PNP, polyneuropathy; QST, quantitative sensory testing; SSR, sympathetic skin response.
The cardiovascular function should be monitored annually by echocardiography, although closer monitoring by electrocardiography every 6 months is recommended, particularly in patients with progressive post-OLT disease. Further assessment of cardiac morphology and function by cardiac magnetic resonance imaging (MRI) and nuclear scintigraphy is also useful for evaluating the progression of cardiac amyloidosis 48 and should be performed annually in the presence of cardiac impairment.
Because some post-OLT patients experience a worsening of peripheral and autonomic nerve symptoms,12,25,29 annual follow-up of post-OLT patients should combine several tests and investigations for evaluating neurological function, including the Neuropathy Impairment Score (NIS), a composite score of polyneuropathy that correlates with disease progression, 49 and neurophysiological tests. The latter should comprise nerve conduction studies in post-OLT patients with large-fiber polyneuropathy or small-fiber tests, such as the Sudoscan®, the quantitative sudomotor axon reflex test, Thermotest, and sympathetic skin response, in the absence of large-fiber damage. Regarding autonomic manifestations, reduced heart rate variability and variations in blood pressure due to orthostatic hypotension are indicative of cardiac autonomic dysfunction,50,51 whereas a reduction in the mBMI can be related to gastrointestinal symptoms affecting nutritional status. 52 In addition, clinical questionnaires such as the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QoL-DN) and the Small-Fiber Neuropathy-Symptom Inventory Questionnaire (SFN-SIQ) should be performed annually to evaluate neuropathy progression, while the Composite Autonomic Symptom Score 31 (COMPASS-31) can be used to further understand the progression of autonomic symptoms. 53
Because OLT does not stop the production of variant TTR in the eye and choroid plexus, ocular and leptomeningeal manifestations can occur in post-OLT patients.29,33 Thus, ophthalmological assessments should also be performed annually to identify ocular complications, whereas head MRI is required on suspicion of central nervous system involvement.
Despite the limited experience concerning the use of TTR stabilizers and TTR mRNA silencers to treat disease progression after OLT, current evidence suggests that post-OLT ATTRv amyloidosis patients could benefit from these novel therapies. In particular, a phase IIIb clinical trial to evaluate the safety and efficacy of patisiran in patients with worsening amyloidosis after OLT suggests that patisiran treatment should be able to halt post-OLT disease progression without causing additional safety issues, 41 which is consistent with our personal experience. Patisiran is the pharmacological agent that has thus far shown the greatest potential for the management of post-OLT amyloidosis progression. Therefore, evidence suggests that patisiran could be considered the treatment of choice in ATTRv amyloidosis patients with disease progression after OLT. However, no specific treatment is yet available for oculoleptomeningeal involvement, which often occurs in ATTRv amyloidosis patients after OLT, with symptomatic treatment as the only therapeutic alternative. Tafamidis is the only approved treatment for ATTRv amyloidosis that can cross the blood–brain barrier, but the concentration is much lower than in the blood, leading to non-stabilized TTR tetramers in the cerebrospinal fluid. 54 Notably, tolcapone can cross the blood–brain barrier more efficiently and stabilize TTR variants, 55 suggesting that this therapeutic agent could provide a benefit in patients with oculoleptomeningeal amyloidosis.
Conclusions
OLT was the first treatment to alter the natural history of ATTRv amyloidosis. However, periprocedural mortality and long-term comorbidities have been major limitations. As described above, patients may experience progressive post-OLT neuropathy and/or cardiomyopathy. In addition, late complications may occur, including ocular symptoms and leptomeningeal complications.
Because post-OLT ATTR amyloidosis patients have typically been excluded from clinical trials, post-OLT disease progression remains poorly understood. To improve the follow-up and clinical management of these patients, we recommend the tests and assessments listed in Table 5. A general evaluation, including orthostatic hypotension, mBMI, PND score, and Coutinho stage, along with laboratory tests assessing liver and kidney function, immunosuppression status, and cardiac biomarkers, should be performed every 6 months. Because progressive cardiomyopathy is the main cause of mortality in post-OLT ATTR amyloidosis patients, assessment of cardiac function by ECG and echocardiography should be performed every 6 and 12 months, respectively. In addition, annual cardiac MRI and nuclear imaging to further assess cardiac morphology and function are recommended in the follow-up of post-OLT patients with cardiovascular impairment. Finally, we recommend the assessment of neuropathy progression via annual neurological and neurophysiological tests.
In a recent study, patisiran demonstrated an excellent safety profile and promising clinical results in post-OLT patients, suggesting that it could be the treatment of choice for progressive ATTRv amyloidosis post-OLT. Tolcapone could, in turn, be considered through extended access for oculoleptomeningeal manifestations because it can cross the blood–brain barrier and stabilize TTR variants.