
Abstract
Granulomatosis or eosinophilic granulomatosis with polyangiitis (GPA/EGPA) can affect multiple organs resulting in heterogeneous symptoms and phenotypes. Pituitary gland dysfunction rarely occurs in GPA (1–3%) and even less in EGPA (two case reports). Here, we report a case of a 51-year-old female patient with a four-year history of EGPA who presented with new polydipsia and polyuria. Laboratory testing and magnetic resonance imaging (MRI) confirmed pituitary gland dysfunction caused by a hypophysitis. Therapeutic adjustment with a switch from dupilumab to mepolizumab resulted in a decrease in clinical symptoms, inflammation in MRI, and normalization of C-reactive protein in serum. This case underlines hypophysitis as a rare organ involvement also in EGPA. Moreover, this case demonstrates the responsiveness of neuroinflammatory manifestations to the recently approved anti-interleukin-5 monoclonal antibody mepolizumab as a new potential treatment option.
Keywords
Introduction
Granulomatosis or eosinophilic granulomatosis with polyangiitis (GPA/EGPA) are multisystem autoimmune diseases characterized by necrotizing, granulomatous small-vessel vasculitis. In case of EGPA, eosinophil-rich inflammation is observed. First symptoms mimic an allergic phase with asthma, allergic rhinitis or sinusitis, while subsequent phases of necrotizing vasculitis can affect multiple organs like the lungs, gastrointestinal tract, or skin and peripheral or cranial nerves. Pituitary gland involvement is reportedly a rare condition in GPA (1–3%).1–4 In EGPA, involvement is even less with only two case reports in the published literature.5,6 Various drug combinations were reported as treatment options, with glucocorticoids and cyclophosphamide being the most common combination.
Case description
A 51-year-old female patient with a four-year history of EGPA presented with new polydipsia and polyuria to our neurological outpatient clinic in a routine checkup. Regarding the disease course, EGPA was initially diagnosed four years ago with mononeuritis multiplex, sinusitis, bronchial asthma, and mastoiditis. Laboratory testing showed positive perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA) (myeloperoxidase (MPO), enzyme immunoassay (EIA)/serum > 1000 U/ml) and increased blood eosinophilia of 30.8% (normal range 0.5–5%). Blood eosinophilia normalized after treatment initiation with cyclophosphamide and rituximab. However, asthma and sinusitis did not fully stabilize under maintenance therapy with methotrexate and prednisolone over a period of two years. Therefore, treatment with the interleukin (IL) 4/13–targeting monoclonal antibody dupilumab was added in addition to background medication with methotrexate and prednisolone, resulting in symptom relief and disease remission for two years. She remained in interdisciplinary outpatient follow-ups in our department and the department of rheumatology. In the current case, polyuria and polydipsia occurred two years after treatment initiation with dupilimumab as new clinical symptoms. We suspected a possible pituitary gland dysfunction regarding polydipsia and polyuria, which was previously described as a rare organ involvement in GPA. After referral to our endocrinologists, laboratory testing and magnetic resonance imaging (MRI) confirmed central diabetes insipidus caused by hypophysitis (Figure 1(a) and (c)), associated with increased levels of C-reactive protein (CRP, 8.6 mg/l). A symptomatic treatment with desmopressin was initiated with two doses per day. Based on laboratory and MRI results, hypophysitis was regarded as a new organ involvement of EGPA, and treatment was adjusted by replacing dupilumab with the anti-IL-5 monoclonal antibody mepolizumab within three months after initial symptom onset. Within four weeks, the patient had improved clinical symptoms of pituitary gland dysfunction, and desmopressin could be reduced to one dose per day. Imaging revealed resolution of inflammation in MRI (Figure 1(b) and (d)), while CRP normalized (Figure 1(e)).
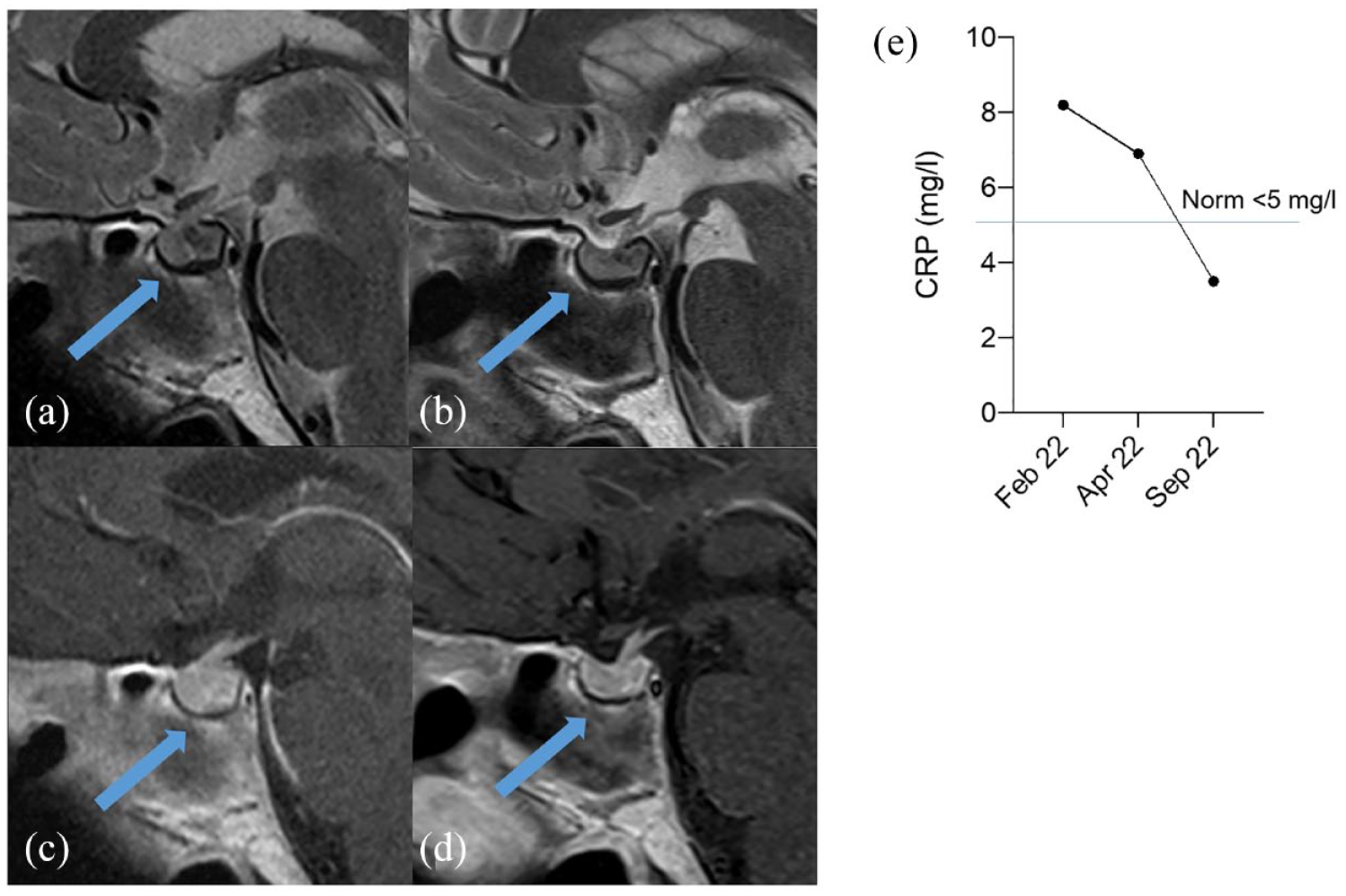
Native sagittal T2 tse (a, b) and post-contrast T1 tse (c, d) sequences of the pituitary gland. The pre-therapeutic scan shows markedly increased volume of the pituitary gland (a, c). The three-month follow-up image shows normalized pituitary volume (b, d). Graph showing C-reactive protein (CRP) normalization after treatment adjustment to mepolizumab (e).
Conclusions
We describe a rare case of hypophysitis in EGPA, developing under treatment with dupilumab that was associated with increased levels of CRP, but not with an exacerbation of other disease manifestations. This case underlines the possible occurrence of new and rare organ involvements like pituitary gland dysfunction even years after disease onset in EGPA. The review of published literature in pituitary gland dysfunction in GPA/EGPA revealed a broad, heterogeneous usage of immunosuppressant agents reaching from oral or intravenously administered corticosteroids, cyclophosphamide, and rituximab to mycophenolate mofetil or even alemtuzumab. 1 Treatment effectiveness varied. Mepolizumab is an anti-IL-5 monoclonal antibody and proved to be an efficacious treatment option in severe asthma.7,8 IL-5 production is pathogenetically central for the development of EGPA by promoting proliferation, transvascular migration, and functional activation of eosinophils. Since 2021, mepolizumab is approved by the EMA for EGPA in a dosage of 300 mg every four weeks based on positive results from the phase 3 clinical trial. 9 Notably, in our case, treatment with mepolizumab was associated with a fast and durable clinical response with a follow-up of eight months, resulting in a complete resolution of hypophysitis by means of imaging with MRI and normalization of CRP. To our knowledge, this is the first case describing mepolizumab as a treatment option for pituitary gland dysfunction in EGPA. This case further highlights the efficacy of mepolizumab, even in rarer disease manifestations like pituitary gland dysfunction related to hypophysitis.