
Abstract
Hepatic vein thrombosis is a rare occurrence in the clinical course of eosinophilic granulomatosis with polyangiitis (EGPA). The major mechanism of thrombosis has been postulated to involve the release of toxic proteins from eosinophils. A 36-year-old man with EGPA was admitted to our hospital in July 2018 with hematemesis and melena. Findings on physical examination included ascites and pigmentation of the lower extremities. Ultrasonography of the hepatic vein and inferior vena cava showed an obstruction of the hepatic vein. Magnetic resonance imaging showed low enhancement in the right hepatic vein region. At 34 years of age, the patient’s EGPA had initially presented as asthma with eosinophilia (white blood cell count of 11.46 × 1012/L with 14.6% eosinophils). His skin biopsy showed infiltration of inflammatory cells and eosinophils, especially around medium-sized vessels, which was consistent with EGPA. The patient was thus diagnosed with Budd–Chiari syndrome associated with EGPA.
Keywords
Introduction
Budd–Chiari syndrome (BCS) is a rare, heterogeneous, and potentially lethal condition characterized by obstruction of the hepatic venous outflow tract. The syndrome is caused by a wide variety of conditions, including congenital or acquired inferior vena cava (IVC) webs and thrombotic, inflammatory, or neoplastic disorders. 1 Eosinophilic granulomatosis with polyangiitis (EGPA) (formerly called Churg–Strauss syndrome) is an eosinophil-rich, necrotizing granulomatous inflammation often involving the respiratory tract, with necrotizing vasculitis predominantly affecting small- to medium-sized vessels. It occurs in people with asthma and eosinophilia. The association between eosinophilia and BCS is related to eosinophilic infiltration of the walls of the IVC, hepatic veins, or both. Massive eosinophil infiltration leads to endothelial cell damage and a hypercoagulable state within the involved vessels. We herein report a unique case of BCS presenting with upper gastrointestinal bleeding, ascites, and hepatic vein thrombosis in association with EGPA.
Case report
A 36-year-old man with EGPA was admitted to our hospital in July 2018 with hematemesis and melena. His EGPA had initially presented at the age of 34 years as asthma with an elevated eosinophil count (white blood cell count of 11.46 × 1012/L with 14.6% eosinophils). There was no evidence of a parasitic infection or an adverse drug reaction to explain the eosinophilia, and the patient was treated with inhaled corticosteroids. Approximately 6 months later, the patient developed a cold, painful, numb, paresthetic right upper extremity and loss of deep tendon reflexes; a cyanotic right finger; and erythematous papules on his extremities (Figure 1). A Doppler sonogram showed thrombi obstructing the right radial and ulnar arteries. Bone marrow biopsy revealed a hyperplasic bone marrow with increased eosinophils, and skin biopsy showed vasculitis with infiltration of inflammatory cells, especially eosinophils, around the medium-sized vessels. The patient was diagnosed with eosinophilic vasculitis (specifically EGPA), which was treated with corticosteroids (prednisolone), thrombolytic therapy (urokinase), and anticoagulation (low-molecular-weight heparin). His peripheral eosinophil count normalized immediately after beginning prednisolone, which was subsequently tapered to 8 mg/day during the following 12 months.

Erythematous papules and maculopapular rash on both lower extremities. (a) Right leg and (b) Left leg.
At the time of the patient’s admission for hematemesis and melena, physical examination revealed ascites, abdominal tenderness, purpura, pigmentation of the lower extremities, and loss of sensation in the distal upper and lower bilateral extremities. His spleen was not palpable, and he had no dilated abdominal wall veins or lower extremity edema. His family history was negative for liver disease. Laboratory data revealed a hemoglobin concentration of 108 g/L, red blood cell count of 3.72 × 1012/L, γ-glutamyl transpeptidase concentration of 244.7 U/L (reference range, 10–60 U/L), albumin concentration of 29.2 g/L (reference range, 40–55 g/L), and normal aspartate aminotransferase and alanine aminotransferase concentrations. His D-dimer concentration was elevated at 604 μg/mL (reference range, 0–232 μg/mL). The concentrations of rheumatoid factor, antinuclear antibodies, antimitochondrial antibody, and proteins C and S were normal. Stool analysis showed no evidence of parasitic infestation, and hepatitis B and C markers, immune indicators, and tumor markers were negative. A chest radiograph and echocardiogram were normal. Gastrointestinal endoscopic examination revealed esophageal varices, and ultrasound of the hepatic vein and IVC showed hepatic venous obstruction, possible compression of the posterior hepatic segment of the IVC, and blockage of the distal hepatic IVC segment. Magnetic resonance imaging of the abdomen showed hepatic morphology consistent with chronic liver damage, peripheral varices of the lower esophagus, a low-enhancing shadow in the area surrounding the right hepatic vein, and an enlarged spleen (Figure 2). Because these findings suggested the possibility of BCS, hepatic venography was performed. This showed segmental development of the middle and right hepatic veins as well as several small hepatic veins joining the third hilar accessory hepatic vein and then entering the IVC. Liver biopsy showed a disordered hepatic acinar structure with dilated central veins and surrounding hepatic sinuses and prominent stasis of red blood cells. These findings were consistent with BCS.
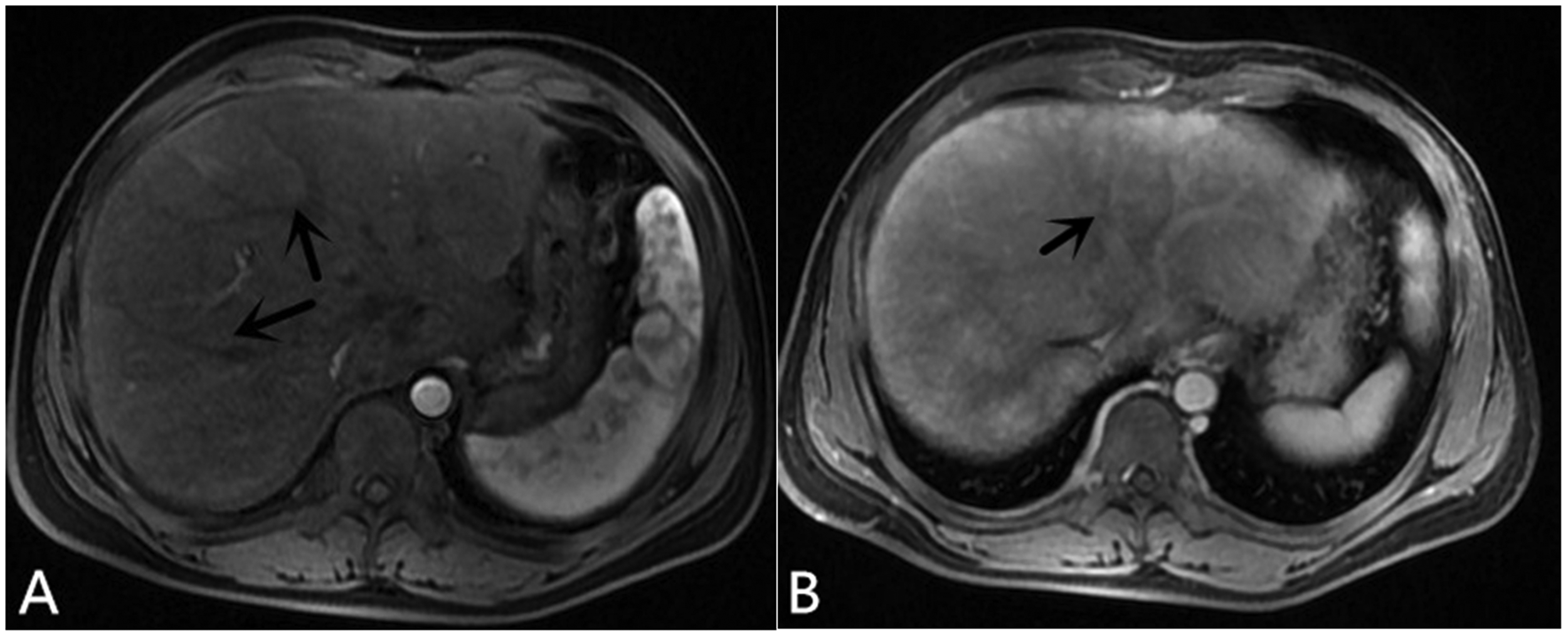
Magnetic resonance imaging showing (a) morphological changes of chronic liver damage and tortuous intrahepatic vessels (arrows) and (b) low-enhancing shadow in the area surrounding the right hepatic vein (arrow).
A transjugular intrahepatic portosystemic shunt was attempted but failed because of complete obstruction of the hepatic veins. The patient was treated with hemostatic drugs and gastric mucosal-protective medications. Diuretic therapy was used to reduce the ascites. During regular follow-up, the patient reported no discomfort, and his eosinophil levels remained normal. A timeline of the leading clinical events is presented in Figure 3.

Timeline of leading clinical events.
Discussion
BCS is a rare but fatal disease resulting from occlusion of the major hepatic veins or suprahepatic IVC. 2 Anatomic abnormalities such as vascular webs or strictures may be present, potentially predisposing to venous thrombosis. 3 The clinical manifestations of BCS depend on the extent and speed of hepatic vein occlusion and the development of venous collateral circulation.
BCS is classified as primary when the hepatic venous outflow obstruction is related to a primary venous problem, such as thrombosis, stenosis, or webs. In the Western world, classical BCS is the most common form of primary BCS. In these cases, the most frequent cause of hepatic vein occlusion is thrombosis secondary to a hypercoagulable state resulting from a thrombophilic disorder, 4 which may be hereditary or acquired. 2 Myeloproliferative neoplasms are present in 35% to 50% of European patients with BCS and are usually associated with the JAK2 V617F mutation. 4 Other causes of BCS include paroxysmal nocturnal hemoglobinuria, antiphospholipid syndrome, and inherited deficiencies of protein C, protein S, and antithrombin III. 5 These conditions are usually combined with underlying thrombophilias. Thus, patients with BCS often have a combination of prothrombotic risk factors.
EGPA was first described in 1951 by Jacob Churg and Lotte Strauss. 6 It is a systemic necrotizing vasculitis of small- and medium-sized vessels and is characterized by asthma, hypereosinophilia, and vasculitis affecting a number of organs. 7 In 1990, the first classification criteria for EGPA were proposed by the American College of Rheumatology. 8 These criteria included six items: (1) asthma, (2) 10% eosinophils on a complete blood count, (3) mononeuropathy, mononeuropathy multiplex, or polyneuropathy, (4) migratory or transient pulmonary opacities detected radiographically, (5) paranasal sinus abnormalities, and (6) biopsy-confirmed extravascular eosinophilic accumulation around a blood vessel. The presence of four or more of these criteria yielded a sensitivity of 85.0% and specificity of 99.7%. 9 Our patient met four of these criteria: asthma, eosinophilia of >10%, peripheral neuropathy, and vasculitis with extravascular eosinophils confirmed by skin biopsy. Asthma is the major manifestation of EGPA, affecting 91% to 100% of patients and often appearing before the systemic vasculitis. Asthma often begins from approximately 30 to 40 years of age, but it may first appear in childhood. Neurologic involvement is the most distinctive feature of the vasculitic process in EGPA. Peripheral neuropathies have a broad range of severity and clinical manifestations because they can affect motor, sensory, and autonomic fibers. Skin lesions are also common in EGPA and include palpable purpura, maculopapular rash, and even subcutaneous nodules, which were observed in 45%, 40%, and 20% of patients, respectively, in one case series. 10 Skin involvement is an important feature and often the site of a positive biopsy.
Our patient developed asthma at 34 years of age and manifestations of systemic vasculitis (including arterial thrombosis) approximately 6 months later. He had various manifestations of peripheral neuropathy, including numbness, paresthesia, pain, weakness, and loss of deep tendon reflexes. The patient had eosinophilia when he initially presented with asthma, and his peripheral blood eosinophil count promptly decreased to normal after initiating corticosteroid treatment. When the patient was admitted to our hospital at 36 years of age, his main manifestations were gastrointestinal bleeding and extensive thrombosis of the portal vein system, which was likely due to eosinophil infiltration of the venous endothelium of intrahepatic vessels. Most tissues and organs can be potentially affected by eosinophilic inflammation in EGPA, including the skin, lungs, gastrointestinal tract, heart, and central and peripheral nervous systems. Both vessel inflammation and eosinophilic proliferation are thought to contribute to organ damage (depending on the target organ), and various clinical effects, including thrombotic complications, can occur. However, the literature contains only a few cases of major thrombotic complications in patients with EGPA. These include a 13-year-old boy with deep vein thrombosis and pulmonary infarction 11 and a 48-year-old man with thrombosis of the left internal iliac and femoral veins. 12 Involvement of the hepatobiliary system in EGPA is very rare but has been reported. 13
The pathogenesis of EGPA remains largely unknown, but abnormal eosinophilic proliferation and tissue toxicity from eosinophil-derived proteins are thought to play major roles. 7 Eosinophils are complex cells that produce an array of molecules with diverse functions. 14 Upon activation, eosinophils release four main granule proteins—major basic protein, eosinophil-derived neurotoxin, eosinophil cationic protein, and eosinophil peroxidase—into the extracellular fluid, where they exert diverse functions. 15 Most importantly, these four proteins can cause hypercoagulation. For example, major basic protein can inhibit the anticoagulant activities of the endothelial membrane by binding to thrombomodulin, 16 and eosinophil cationic protein can stimulate reactions that are dependent on the presence of factor XII. 17 Hypothiocyanous acid, the main oxidation product of eosinophil peroxidase, has been shown to stimulate tissue factor expression, thereby promoting thrombosis. 18 Therefore, eosinophil-derived factors may promote thrombogenicity of the endothelial lining, stimulate platelet activation, and increase the synthesis of hemostatically active plasma proteins, favoring a hypercoagulable state. Furthermore, tissue damage by eosinophils may also contribute to thrombosis. Thus, considering the above-mentioned components, massive eosinophil infiltration can lead to damage of the endothelium and a hypercoagulable state. 19 It is likely that elevated eosinophils play an important role in the pathogenesis of hepatic venous thrombosis.
Systemic vasculitides are clinically diverse, multisystemic diseases with the common pathological feature of inflammation of blood vessel walls. This inflammation may in turn predispose to vascular thrombosis. Vasculitis might trigger thrombosis through inflammation, inflammatory cytokines, and other molecules involved in vessel wall injury. 12 More specifically, eosinophilic inflammation may be a risk factor for thrombotic events. We hypothesize that a patient’s long-term outcome is influenced by his or her underlying vascular disorder and that hepatic vein thrombosis is generally considered a rare occurrence in the course of EGPA.
It is therefore essential to consider the diagnosis of BCS in all patients with thrombophilia. Activated eosinophils in patients with EGPA can provoke thrombosis in various organs, and the case presented here suggests that the thrombosis affected the portal venous system, producing BCS.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
The Independent Institutional Review Board of the First Hospital of Jilin University approved the study protocol. The patient described in this report provided verbal informed consent.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Science and Technology Development Program of Jilin Province (Grant no. 20190103079JH) and the Youth Development Foundation of the First Hospital of Jilin University (Grant no. JDYY102019004).