
Abstract
Lipid storage myopathies (LSMs) are metabolic disorders of the utilization of fat in muscles due to several different defects. In this review, a molecular update of LSMs is presented and recent attempts of finding treatment options are discussed.
The main topics discussed are: primary carnitine deficiency, riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency, neutral lipid storage disorders and carnitine palmitoyl transferase deficiency. The most frequent presentations and genetic abnormalities are summarized. We present their diagnosis utilizing biomedical and morphological biomarkers and possible therapeutic interventions. The treatment of these metabolic disorders is a subject of active translational research but appears, in some cases, still elusive.
Introduction
Lipid storage myopathies (LSMs) are a heterogeneous group of genetic disorders characterized by pathological lipid accumulation in muscle fiber. They are characterized on morphological grounds by the accumulation of lipid droplets (LDs) in muscle [Figure 1(a and b)] as well as in other tissues, due to several biochemical or molecular defects (Table 1).

(a) Muscle biopsy in a patient with lipid storage myopathy and mitochondrial abnormalities (trichrome stain). (b) Lipid myopathy (Sudan black b stain).
Classification of lipid myopathies.

There are four types of diagnosable LSMs that always present with an abnormal storage of neutral lipids typically in muscle: primary carnitine deficiency (PCD), multiple acyl-CoA dehydrogenase deficiency (MADD), neutral lipid storage disease with ichthyosis (NLSDI), and neutral lipid storage disease with myopathy (NLSDM).1–3 Carnitine palmitoyl transferase (CPT-II) deficiency is not classically considered as an LSM, but it was included in this review, as, in some patients, an accumulation of neutral lipids inside muscle LDs has been detected. 4 Some LSMs can be managed by an appropriate diet and cofactor supplementation. For example, PCD patients show a dramatic improvement with high doses of oral L-carnitine supplementation, while most patients with late-onset MADD are responsive to treatment with riboflavin and CoQ supplementation.
CPT-II deficiency 5 and carnitine deficiency syndrome 6 were identified on biochemical grounds. In carnitine systemic primary deficiency syndrome, an OCTN2 pathogenetic mutation was identified in the patient described by Chapoy and colleagues, 7 as well as in other cases. 8 The carnitine deficiency syndrome is characterized by myalgia, fluctuating weakness and hypotonia with cardiomyopathy. MADD is an increasingly recognized entity characterized by proximal myopathy with limb and neck muscle weakness. In MADD, symptoms and age onset are highly variable and characterized by recurrent episodes of lethargy, vomiting, hypoglycemia, metabolic acidosis and hepatomegaly, often preceded by a metabolic stress. MADD is also known as ‘glutaric aciduria type II’, because it results in the large excretion of glutaric, lactic, ethylmalonic, butyric, isobutyric, 2-methylbutyric and isovaleric acids. MADD can be caused by mutations in three different genes (ETFA, ETFB, ETFDH); however, in most patients the disease is caused by mutations in the ETFDH gene, encoding electron transfer flavoprotein dehydrogenase (ETFDH). 9
Finally, neutral lipid storage disease (NLSD) includes an heterogeneous group of metabolic disorders (NLSDI and NLSDM) characterized by an accumulation of triglycerides contained in cytoplasmic LDs of several tissues, including the skin, muscle, liver, bone marrow, and intestine.
NLSDI or Chanarin–Dorfman syndrome (CDS) is characterized by ichthyosis associated with mild myopathy and spleno hepatomegaly with various ophthalmologic symptoms (cataract, nystagmus, strabismus), hearing loss, mild mental retardation, short stature, microcephaly and intestinal involvement.10–12 Typically, individuals with CDS show multiple vacuolizations of the neutrophilic and eosinophil leukocytes (Jordans’ anomaly) in peripheral blood smears. 13 In CDS, muscle abnormalities have been detected in almost 40% of patients. Myopathy typically begins in the 30s, but it has also been described in young children.12,14–16 NLSDI is caused by a molecular defect in the ABHD5 gene, which codifies for the activator of adipose triglyceride lipase (ATGL).
NLSDM mainly has a myopatic presentation with asymmetric distal limb weakness. In patients with NLSDM, triglyceride accumulation is due to an ATGL deficiency. This enzyme catalyzes the first step in the hydrolysis of fatty acids from triacylglycerol stored in the LDs.
LSMs, impairing energy production, always involve skeletal muscle and cause progressive myopathy with muscle weakness, or recurrent acute episodes of rhabdomyolysis triggered by exercise, fasting, or infections. 4 Molecular characterization of these disorders has important implications both for accurate diagnostic approach and for development of therapeutic strategies. The treatment of these metabolic disorders is a subject of active translational research but appears in some cases still elusive (Table 2).
Therapy of lipid myopathies.

ACAD9, acyl-CoA dehydrogenase 9 deficiency; CACT, carnitine acylcarnitine translocase deficiency; CPT-II, carnitine palmitoyl transferase II deficiency; DECR, dienoyl-CoA reductase deficiency; LCAD, long-chain acyl-CoA dehydrogenase deficiency; LPN1, phosphatidic acid phosphatase deficiency (lipin1 deficiency); MADD, multiple acyl-CoA dehydrogenase deficiency; MCAD, medium-chain acyl-CoA dehydrogenase deficiency; MCKAT, medium-chain 3-ketoacyl-CoA thiolase; MCT, medium-chain triglyceride; MTP, mitochondrial trifunctional protein deficiency; NLSDI, neutral lipid storage disease type I; NLSDM, neutral lipid storage disease type M; PCD, primary carnitine deficiency; SCAD, short-chain acyl-CoA dehydrogenase deficiency; SCHAD, short-chain L3-hydroxyacyl-CoA dehydrogenase deficiency; VLCAD, very-long-chain acyl-CoA dehydrogenase deficiency.
Clinical presentation and pathogenesis of LSMs
Primary carnitine deficiency
PCD syndromes are rare biochemical disorders and can be classified on the basis of clinical and biochemical criteria in muscle carnitine deficiency (OMIM#212160) and systemic carnitine deficiency (OMIM#212140). A carnitine deficiency syndrome should be suspected in a patient with LSM when the following symptoms are present: myalgias, fluctuating weakness, hypoglycemia, with or without ketoacidosis with a Reye-like syndrome, abnormal fatigability, cardiomyopathy with left axis deviation.17,18 Primary systemic carnitine deficiency is a well-recognized childhood treatable entity characterized by progressive cardiomyopathy, LSM, attacks of hypoglycemia, and hepatomegaly with a Reye-like syndrome that may lead to permanent brain damage. In several cases, a defect of the carnitine ‘high-affinity’ transport organic cation transporter 2 (OCTN2) protein has been demonstrated in cultured fibroblasts. 19 OCTN2, coded by the SLC22A5 gene, is composed of 12 transmembrane domains (TMDs), both N- and C-terminal regions facing the cytoplasm and an extracellular loop located between the first two TMDs. This loop includes three residues, N57, N64, and N91, that play a key role in substrate and sodium recognition. The amino terminal part and the region placed between TMD7–11 are involved in carnitine recognition while TMD7–12 are Na+/carnitine complex transport regions. Finally, a glucose-transporter motif is located between TMD2 and TMD3 [Figure 2(a)].20,21 Until now, about 150 variations of the SLC22A5 gene have been identified; the majority of them are missense mutations. 22 Some functional studies demonstrated that nonsense and frameshift mutations lead to a loss of protein function, while missense substitutions affect the normal localization of OCTN2 on the plasma membrane or reduce carnitine recognition and transport. 22 In particular, variations of N57, N64, and N91 can cause cytoplasmic retention of OCTN2, while variations that affect the other residues of the extracellular loop can result in an impairment of carnitine recognition. 21

Domains organization of proteins involved in LSM onset and of CPT2 protein. (a) The OCTN2 protein is composed of 557 amino acids organized in 12 TMDs and a nucleotide binding motif. The region between N-terminal and TMD-5 is involved in carnitine and sodium recognition while the C-terminal part contains carnitine recognition (TMD7–D11) and transport sodium-dependent (TMD7–12) regions. The protein also has three putative glycosylation sites (N57, N64, and N91) and six potential sites for protein kinases (S164, S225, S280, S322, S323 and S402). Moreover, a glucose-transporter signature motif is located between TMD2 and TMD3; (b) the ETFDH protein, consisting of 617 amino acids, comprises three functional regions: the 4Fe4S cluster; the FAD-binding domain; the UQ-binding domain. Furthermore, ADP-binding motif is located in the FAD domain and two membrane-binding surface regions are contained in the UQ domain; (c) schematic representation of ATGL (amino acids 1–504) shows a patatin domain that contains the catalytic site (residues S47 and D166) and a LIR motif. In the C-terminal part, there is a hydrophobic domain involved in LD binding; (d) ABHD5 protein (amino acids 1–349) has an LD-binding region (hydrophobic domain) and an α&x47;α hydrolase domain that includes two residues involved in ATGL interaction (Q130 and E260); (e) CPT2 (amino acids 1–658) has two functional sites: a mitochondrial membrane-binding domain and a long acyl groups-binding site. Moreover, H372 represents the catalytic site.
Riboflavin-responsive multiple acyl-CoA deficiency
MADD (OMIM#231680) are multisystem genetic diseases characterized by various clinical manifestations with different degrees of severity. The most common clinical phenotype is the type III (RR-MADD), often associated with ETFDH mutations. ETFDH, also called electron transfer flavoprotein (ETF)-ubiquinone (UQ) oxidoreductase, is a protein localized in the inner membrane of mitochondria, playing a central role in the electron-transfer system. 23 Indeed, ETFDH mediates electron transport from flavoprotein dehydrogenases to the ubiquinone pool. 24 ETFDH protein consists of 617 aa residues and possesses three functional regions: flavin adenine dinucleotide (FAD)-binding domain, 4Fe4S cluster and UQ-binding domain (Figure 2b). An ADP-binding motif is also localized in the FAD-binding domain (aa residues G42-G47) and two membrane-binding regions are identified within the UQ-binding domain (aa residues F114-L131 and G427-W451).
Until now, about 700 RR-MADD patients have been reported all over the world.25–29 A total of 640 (95%) were affected by MADD type III. Molecular analysis was performed for 523 of these patients and 187 different mutations were identified in the ETFDH gene [Figure 3(b)]. The majority of alterations are missense mutations (73%): 1% occur in the N-terminal region, 12% in 4Fe4S cluster, 50% in the FAD domain, 31% in the UQ domain and 6% in the C-terminal region [Figure 3(c)]. The remaining variations are frameshift mutations (13%), splice site variations (8%) and nonsense mutations (6%). All patients presenting MADD type III carry at least one missense variation (Table 3). It is well documented that many missense mutations impair FAD binding.30,31 FAD plays a central role in the promotion of conformational stabilization and correct folding of many flavoproteins. 32 An increase in FAD concentration may restore the stability of most of the ETFDH proteins carrying missense mutations. Some in vitro studies have been performed using fibroblasts obtained from MADD patients to test the stability and activity of ETFDH.30,33 In particular, fibroblasts of patients carrying different missense mutations of ETFDH (p.P456L, p.P483L, and p.G429R), cultured with high concentrations of riboflavin in the medium, showed increased protein stability. 30 These variants lead to a milder impairment of the native folding of ETFDH and the treatment with riboflavin partially restores enzymatic activity, although does not replace conformational alterations. MADD is also associated with the mutations in ETFα, ETFβ, SLC52A1, and FLAD1, although rarely.25,34–37

Clinical phenotype of MADD patients and mutations identified in the ETFDH gene. (a) Percentage of MADD type I, type II and type III in about 700 patients; (b) Frequency of ETFDH different variations found in 523 late-onset MADD patients; (c) Distribution of missense mutations in ETFDH protein regions.
Genotype and response to Riboflavin.
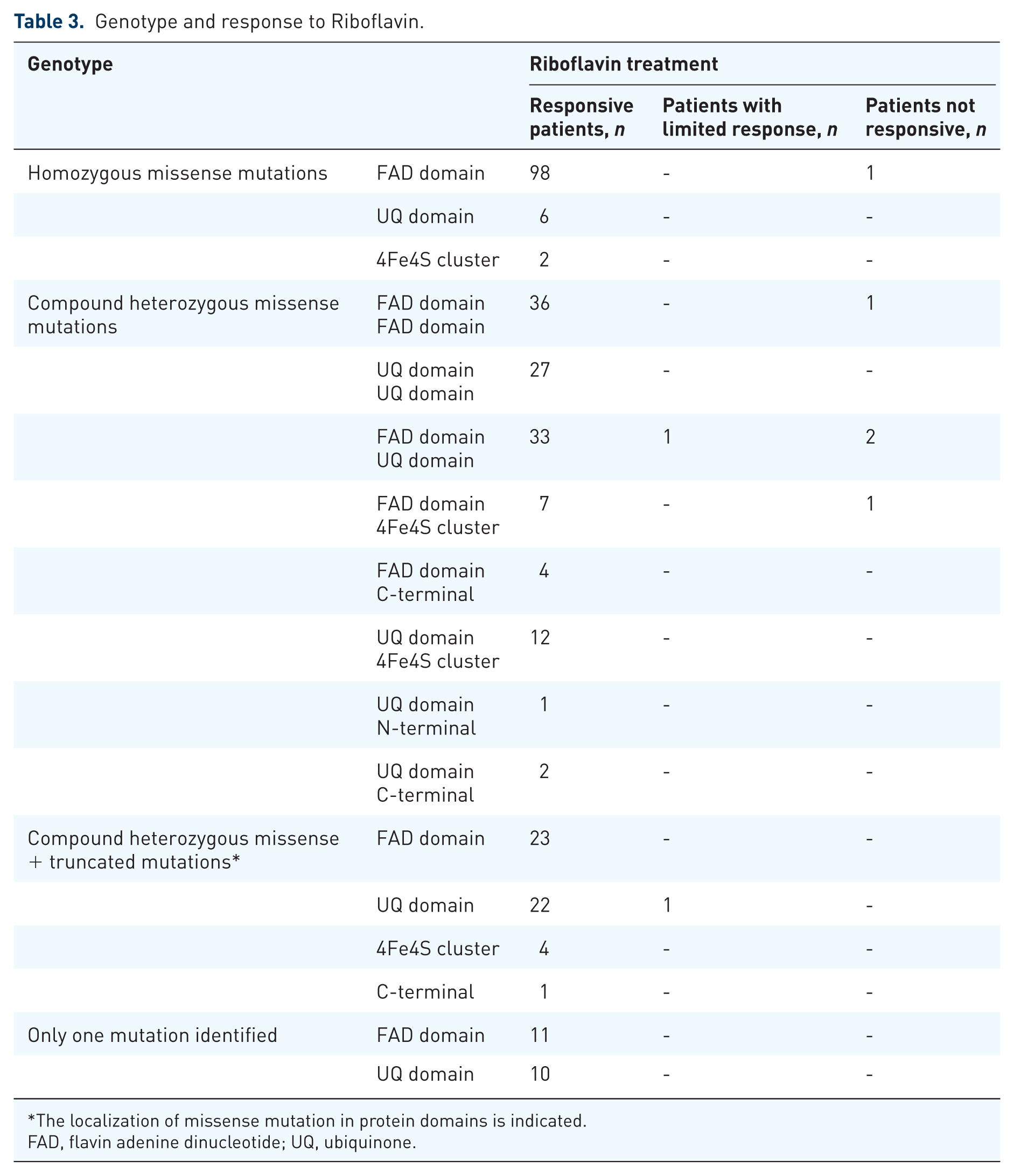
The localization of missense mutation in protein domains is indicated.
FAD, flavin adenine dinucleotide; UQ, ubiquinone.
Neutral lipid storage disorder
NLSD is a heterogeneous group of inherited disorders, consisting of two autosomal recessive diseases: NLSDM and NLSDI.
Mutations in the PNPLA2 gene cause the onset of NLSDM (OMIM#610717). 38 PNPLA2 codifies for the ATGL lipase. Human ATGL consists of 504 amino acids and contains a patatin domain in a three-layer sandwich architecture (αβα) located within the NH2-terminal region of the protein. The catalytic site is located in the patatin domain and is characterized by a catalytic dyad (amino acid residues: S47 and D166). The COOH-terminal region of ATGL contains a stretch with lipase regulatory activity and also mediates LD binding [Figure 2(c)]. ATGL catalyzes the first step in the hydrolysis of triacylglycerols (TAGs) generating diacylglycerols (DAGs) and free fatty acids (FAs). The enzyme exhibits high substrate specificity for TAGs and only low activity for DAGs and phospholipids. DAG is the major substrate for hormone-sensitive lipase activity, which determines the production of FA and monoacylglycerol (MAG). Finally, monoglyceride lipase (MGL) catabolizes MAG to FA and glycerol. Lipids generated from these processes are major energy substrates, as well as precursors of membrane and signaling molecules. 35 To date, a total of 37 different ATGL mutations have been reported. Of these, 20 (57%) resulted in premature stop codons and determined the formation of truncated proteins,34,39–53 and 6 (13%) were null mutations leading to the complete absence of ATGL protein production.47,54–56 Finally, 11 variations (30%) were missense mutations.45,47,48,55,57–60 All molecular data collected on NLSDM families suggest a marked genetic heterogeneity for this disease.49,53,55,59–61
The cause of NLSDI (OMIM#275630) has been attributed to mutations in the ABHD5/CGI-58 coding gene. 62 The ABHD5 protein, a member of the esterase, lipase and thioesterase subfamily, is a 349 amino acid-long protein characterized by the lipid-binding motif and the hydrolase domain that contains Q130 and E260, the essential residues for ATGL interaction [Figure 2(d)]. 63 Indeed, ABHD5 plays a key role in TAG metabolism; it binds to perilipin, localizes to adiposomes and promotes the activation of ATGL. To date, 128 CDS patients were reported. For 85 of these patients, clinical diagnosis has been confirmed by ABHD5 mutation analysis. Overall, 80% of ABHD5 mutations determine the production of truncated proteins, most of which are missing a large portion of the protein structure and the E260 amino acid residue, which allows ATGL interaction.15,64,65 In summary, molecular and genetic data indicate that the ABHD5 protein function is totally lost in most patients with CDS. Cellular ABHD5 triglyceride hydrolase activity is due to the specific activation of ATGL function. However, ATGL is able to hydrolase triglycerides also in the complete absence of the ABHD5 protein, although with low or very high efficiency depending on the type of tissue. 66
Unlike patients with NLSDM, those with NLSDI do not develop cardiomyopathy, as their cardiomyocytes have a smaller but sufficient amount of energy from basic ATGL activity.
CPT-II deficiency: clinical presentation and pathogenesis
The human CPT2 gene codifies for the CPT-II mitochondrial enzyme, a homotetrameric complex located on the inner mitochondrial membrane. CPT-II forms the carnitine palmitoyl transferase (CPT) system together with CPT-I, which plays a key role in the transfer of long-chain FAs from the cytosol to the mitochondrial matrix, where FA oxidation occurs [Figure 2(e)]. Mutations in the CPT2 gene cause the CPT-II muscle deficiency (OMIM#255110), the most common form of muscle FA metabolism disorder. 67 More than 350 patients have been described all over the word. The CPT-II muscle deficiency is an autosomal recessive disorder. To date, different mutations have been identified in the CPT2 gene, homogeneously distributed along the five coding exons with the exception of the p.S113L mutation, which is present in 90% of patients with homozygous or heterozygous status.68–71 Biochemical studies have demonstrated that none of the patients show a complete loss of CPT-II protein72,73, thus indicating that CPT-II patients with the muscular form carry at least one CPT2 allele with a missense mutation. This condition might ensure partial CPT activity in patient tissues.
In the most typical presentations, CPT-II deficiency is seen in young adults experiencing episodes of muscle pain and rhabdomyolysis triggered by prolonged exercise, fasting, cold, or a combination of these. The attacks are associated with pain, stiffness without cramps, and highly elevated creatine kinase (CK) levels (about 50,000 U or maybe even up to 200,000 U) reflecting muscle necrosis. This may lead to acute renal failure.
CPT-II deficiency is routinely diagnosed by the determination of enzyme activity in muscle biopsies by the conversion of (14C) radiolabeled palmitoyl carnitine substrate by the isotope-exchange assay. The diagnosis can be made also on the basis of genetic analysis and acylcarnitine plasma profile or urine.
Biomarkers
The mean biomarkers for PCD are: low free carnitine and total carnitine in plasma and urine, high CK. For RR-MADD, the characteristic glutaric aciduria type II pattern might be found by mass spectrometry during fasting or metabolic crisis. 74
ATGL deficiency and ABHD5 deficiency are suspected by the Jordans’ anomaly in peripheral blood leukocytes or bone marrow megacaryocytes (white blood cells precursors). 61 By this morphological tool both NLSDI and NLSDM has been diagnosed, and screening for genomic DNA mutation is then indicated.
Analysis of acylcarnitine in the blood, using tandem mass spectrometry, was developed in the late 1980s. Only a small amount of plasma (100 μl) or blood spotted into filter paper (Guthrie card) is required, allowing the diagnosis of several inborn errors of FA metabolism. CPT-II deficiency leads to an increase of serum palmitoyl carnitine (C16:0) and oleoylcarnitine (C18:1), a characteristic profile of blood acylcarnitines 75 , whereas short and medium-chain acylcarnitines might be normal during myoglobinurie attacks, free carnitine is low. The plasma and urinary acylcarnitine profile has demonstrated its high value as a fast and non-invasive method for the elevated detection of inborn errors of FA oxidation in the screening of newborns.
It is important to collect samples from patients during the crisis since a nonsignificant profile can be observed in patients during intercritical periods. Overnight fasting is useful but may lead to unexpected hypoglycemic episodes and sudden death in neonatal and infantile forms.
It is noteworthy that patients with CPT-II show the same elevated long-chain acylcarnitine profile in plasma as carnitine/acylcarnitine translocase (CACT) deficiency. These two disorders can be distinguished by their clinical presentation. Only the severe clinical presentation of hepato-cardio-muscular form of CPT-II deficiency can overlap with that of CACT deficiency. Most patients with CPT2 have rhabdomyolytic episodes and direct measurement outcomes of enzymatic activity are required to differentiate between these two metabolic defects. Tandem mass spectrometry of serum acylcarnitines is a rapid screening test that should be included in the diagnostic work-up of patients with recurrent myoglobinuria or cases with high CK and diffuse myalgia and cramps. In particular, in young children suspected of CPT-II deficiency, one could avoid performing invasive muscle biopsy by appropriate acylcarnitine studies.
Treatment guidelines
PCD
Carnitine supplementation corrects cardiomyopathy and other clinical signs.7,17 In some cases, this treatment may prevent the need for cardiac transplantation. 76 The L-carnitine dose may vary from 100 to 600 mg/kg per day on the basis of the calculated carnitine depletion from muscle, liver, heart, and kidney. Individually adjusted dosing may require several plasma level measurements. 74 Side effects for L-carnitine supplementation are diarrhea or a fishy body odor. 77 In some cases, a medium-chain triglyceride (MCT) diet may be added. 78 Muscle and plasma carnitine deficiency is found in primary muscle carnitine deficiency if the clinical syndrome is confined to skeletal muscles6,79; the clinical features are episodes of fluctuating muscle weakness, affecting mostly limb and neck muscles. 74 The patients show appropriate ketogenesis on fasting and on a fat-rich diet. Biochemical features are low muscle carnitine (below 15%) and absence of organic aciduria. Carnitine concentrations in plasma and liver are normal. There is in vitro stimulation of labeled palmitate oxidation by L-carnitine, and oleate. 6 Although much is known about the mechanisms of high-affinity carnitine transport mediated by the OCTN2 transporter, data on muscle-specific transport (low affinity) in human muscle carnitine deficiency cases are still scanty. In a childhood case, abnormal low-affinity carnitine transport 79 was found in cultured muscle cells. This could be due to either slow maturation or an abnormal sarcolemmal carnitine transporter. Muscle carnitine deficiency could be caused by an abnormal low-affinity carrier or by a low number of carriers. It is distinguished from carnitine insufficiency because of the absence of acylcarnitines elevation in plasma or urine. 74 Treatment with an L-carnitine replacement and MCT diet has been successful in a number of cases. 80
The use of carnitine is resolutive in cases of PCD due to OCTN2 deficiency for cardiomyopathy and seems to be useful in secondary carnitine deficiency states or carnitine insufficiency associated with oxidative phosphorylation disorders.77,80,81 The dose of carnitine can reach 3–5 g, in four daily doses, while, in other diseases causing a secondary defect, the recommended dosage is about 100 mg/kg.
RR-MADD
Riboflavin supplementation often results in a marked improvement of clinical features (50–100 mg three times daily). A low fat diet avoiding long fasting periods can also be helpful. A total of 449 patients with ETFDH mutations were treated with riboflavin and 442 of them clearly resulted in a response to therapy.26,28,82–84 In Table 2, the association between genotype and riboflavin response is reported for 334 patients. The majority of these patients were homozygotes (113) or compound heterozygotes (134) for missense mutations. It was not possible to report the genotype for 117 patients who showed a good response to riboflavin treatment83,85 because the authors do not report molecular analysis results for each patient, but only a list of mutations identified in all MADD patients.
Unfortunately, the supplementation was partially or totally not effective in seven patients. Failure to response to treatment is probably due to the presence of mutations that dramatically reduce ETFDH stability or discontinuous therapy started later in life.32,86,87
In order to obtain a better diagnosis, functional studies should be performed to clarify the pathogenic effects of each ETFDH missense mutations identified in patients. Furthermore, early riboflavin treatment should be started in patients with late-onset MADD to prevent severe metabolic crisis.
MADD can be responsive to riboflavin supplementation.
NLSDI
Treatment for NLSDI is symptomatic. Liver abnormalities can occur in more than 80% of patients, ranging from hepatomegaly or liver steatosis to cirrhosis. Since 1980, it was reported that MCT supplementation with a low fat diet (specifically low in long-chain FAs and minimal saturated fat), decreases the liver size and normalizes hepatic enzymes. 12 Many others case reports stated similar results, especially when an early initiation of a low fat diet was started in combination with vitamin E and ursodeoxycholic acid.14–16,65,88 A skin involvement is present in NLSDI in 100% of patients, consisting of a nonbullous congenital ichthyosiform erythroderma. To alleviate skin symptoms, it is recommended to apply emollient cream for local applications. Areas of hyperkeratosis generally respond well to topical retinoids. Some improvement of ichthyosis and mild reduction in aminotransferase levels have been observed in few cases following oral treatment with retinoids89,90, but there is no consensus about the use of retinoids in CDS with abnormal liver function. However, improvement in skin erythroderma without exacerbation of liver abnormalities has been reported with the use of acitretin. 89
NLSDM
At present, NLSDM has no specific therapy and patients develop a severe, irreversible muscle atrophy. Moreover, about 50% of NLSDM patients also develop cardiomyopathy.
An MCT diet and oral carnitine have been used with some improvement 91 but these findings were not confirmed in other studies. The peroxisome proliferator-activated receptor (PPAR)-α agonists are drugs used to reduce TGs in serum and mobilize lipids from LDs. 92 The cardiomyopathy of ATGL-deficient mice resolved following treatment with the PPAR-α agonist Wy14643. 93 PPAR transcription factors regulate the expression of many genes related to lipid and energy metabolism. PPAR agonist treatment is not universally effective in patients with NLSDM. In recent years, few reports describing an improvement of myopathic symptoms using bezafibrate (a PPAR-α receptor agonist) have been published60,94–96, therefore, further studies using bezafibrate are warranted.
Also β-adrenergic stimulation could be used to reduce LDs. 45 In a cellular model therapy clenbuterol, but not salmeterol, significantly reduced LDs. 45
In case of severe and arrhythmogenic cardiopathy, the placement of a defibrillator is mandatory. Finally, an important role is played by physical exercise by activation of alternative way of lipid metabolism and motor rehabilitation.
CPT-II
The main precautionary guideline in LSM due to defects of mitochondrial β-oxidation is the avoidance of fasting. Since patients with metabolic disorders cannot utilize FAs by β-oxidation, the accumulation of toxic intermediate metabolites (i.e. acyl-CoA) should be avoided as soon as the development of the critical signs occurs. In the diet, fat consumption should be restricted to 25% of total calories, and the amount of long-chain FAs (LCFAs) should be minimal. Increased caloric intake from carbohydrates may be necessary during intermittent illness attacks because of increased metabolic request. A low fat, high carbohydrate diet is beneficial in reducing the frequency of rhabdomyolytic attacks in several disorders of fatty acid metabolism, including very-long-chain acyl-CoA dehydrogenase deficiency (VLCAD) and CPT-II deficiency. The current dietary treatment of LCFA defects (high carbohydrates with medium even-chain triglycerides and reduced long-chain fats) is based on evidence provided by expert opinion alone or from descriptive case series without controlled trials. It is difficult to perform double-blind studies to prevent cardiomyopathy, rhabdomyolysis, and muscle weakness. CPT-II myoblasts and VLCAD fibroblasts, treated with fibrates, showed an increase in FA oxidation.97,98 Unfortunately the use of bezafibrate in patient management was proposed as beneficial but not confirmed. 95
A diet high in carbohydrates improves exercise tolerance in patients with CPT-II deficiency. 99 The effect of a high versus low-carbohydrate diet on exercise tolerance has been tested in four patients with CPT-II, who cycled at a constant workload of 50% of VO2max. Frequent meals with high carbohydrate intake, especially before and after prolonged physical activity, improved exercise tolerance in these patients. 100
Defect of β oxidation
As in other metabolism disorders, therapy in patients with FA oxidation defects can be divided into two main stages: acute phase and long-term treatment. In all patients presenting acute clinical pictures with severe hypoketotic hypoglycemia (Reye-like syndromes) or myoglobinuria, the treatment is aimed at stopping the endogenous lipid catabolism responsible for the symptoms. The intravenous infusion of concentrated glucose solutions, eventually associated with insulin therapy, is the only instrument capable of suppressing lipolysis; in cases where there is a lack of carnitine, primitive or secondary, it is also indicated the replacement treatment always intravenously. If there is a coma with severe hepatic impairment that does not respond to conservative therapy, dialytic treatment may be indicated to remove accumulated toxic substances. In LCFA oxidation defects, the use of carnitine is still debated both for the scarce effect 101 and for the danger of a cardiotoxic effect caused by long-chain acylcarnitines. Fibrates are not useful for VLCAD. 102
The long-term treatment of patients with impaired β oxidation is based on some fundamental principles:
avoid prolonged fasting and excessive muscular effort;
establish a diet therapy based on fractionated meals rich in complex carbohydrates and low in fat;
where it can be used and depending on the enzymatic defect, riboflavin and carnitine therapy. 103
Diet therapy may include the use of raw corn starch, as in some forms of glycogen storage,104,105 and the use of MCTs for defects in LCFA metabolism.4,12,106 The primary goal of treatment is to prevent episodes of metabolic decompensation; during follow up, patient monitoring is based on the combination of clinical, biochemical and instrumental evaluations.
Conclusion
An extensive review of literature describing patients with PCD, MADD, and CPT-II reveals that these defects are rare but treatable. Although, even when treated, patients with CPT-II and MADD might present episodes of transient weakness and myalgia. The treatment of NLSDs is still elusive and will need further translational research.