
Abstract
The current case report describes the clinical, biochemical and genetic characteristics of carnitine-acylcarnitine translocase deficiency (CACTD) in infant male and female twins that presented with symptoms shortly after elective caesarean delivery. The clinical manifestations were neonatal hypoglycaemia, arrhythmia and sudden death. The age of onset was 1.5 days and the age of the death was 1.5–3.5 days. Dried blood filter paper analysis was used for the detection of acylcarnitine. Peripheral venous blood and skin samples were used for next-generation sequencing. The twins and their parents underwent gene analysis and whole exome sequencing analyses of the solute carrier family 25 member 20 (SLC25A20; also known as carnitine-acylcarnitine translocase) gene. Both infants carried compound heterozygous variants of the SLC25A20 gene: variant M1:c.706_707insT:p.R236L fs*12 and variant M2:c.689C>G:p.P230R. The M1 variant was paternal and had not been previously reported regarding CACTD. The M2 variant was maternal. CACTD has severe clinical manifestations and a poor prognosis, which is manifested as hypoketotic hypoglycaemia, hyperammonaemia, liver function damage and elevated creatine kinase.
Introduction
Carnitine-acylcarnitine translocase deficiency (CACTD; OMIM212138) consists of a series of energy metabolism disorders caused by the inability of long-chain acylcarnitine to enter the mitochondrial inner membrane and to participate in β-oxidation due to carnitine acylcarnitine translocase deficiency; and it involves vital organs such as the heart, liver and skeletal muscle. 1 The disease is a rare autosomal recessive disorder with an incidence of 0.2–1.8/100 000 reported in the literature. 2 It has a higher incidence in East Asia than in other regions. 3 It has an unknown incidence in mainland China but an incidence of 1/60 000 in Hong Kong. 4 It is reported that the majority of patients with CACTD have a poor prognosis, with a mortality rate of 65%; 1 and approximately 82% of patients develop the disease during the neonatal period. 5 The definite pathogenic variants of the solute carrier family 25 member 20 (SLC25A20; also known as carnitine-acylcarnitine translocase) gene are mainly missense and deletion variants; and different variants result in different degrees of reduction of carnitine-acylcarnitine translocase activity and different clinical phenotypes. 6 Severe cases generally die shortly after birth and mild cases survive to 16 years with early treatment. 7 Early detection of this disease is achieved by measuring acylcarnitine in dried blood on a filter paper.
This current case report describes the clinical, laboratory and genetic characteristics of two newborns, male and female twins, with CACTD. Both infants carried SLC25A20 gene compound heterozygous variants inherited from both of their parents.
Case report
On 28 November 2021, a G1P2 male infant (gestational age 37 + 6 weeks; birth weight 3490 g) and a G1P2 female infant (gestational age 37 + 6 weeks; birth weight 3490 g) were admitted to Fenghua District People's Hospital, Ningbo, Zhejiang Province, China, for their mother to have an elective caesarean section because she was carrying twins. The amniotic fluid was clear and the umbilical cords as well as the placentas were unremarkable. There were no fetal membranes and the Apgar scores for both infants at birth were normal. The family history revealed healthy parents with a non-consanguineous marriage. Family members denied a history of diseases such as ‘hepatitis’ and ‘tuberculosis’. After a full-term caesarean section, the two infants developed symptoms that included a poor response, hypoglycaemia, hypotonia, arrhythmias and sudden cardiorespiratory arrest on day 1.5. Both infants were transferred to the Newborn Centre, Ningbo Women and Children’s Hospital, Ningbo, Zhejiang Province, China due to ‘facial cyanosis for 6 h and 1 h after cardiopulmonary resuscitation’. The female infant died on the way to the hospital due to more severe symptoms and a poor response to treatment so there were no further investigations undertaken other than a genetic analysis from a skin sample. The vital signs of the male infant at admission were as follows: temperature, 36.0°C; heart rate, 88 beats/min (bpm); weak spontaneous breathing; blood pressure, 68/40 mmHg; transcutaneous monitoring of arterial oxygen saturation of haemoglobin, 65% (T combination); no reaction; weak spontaneous and wheezing breathing; flat and soft anterior fontanel; soft neck; decreased lung sounds in both lungs; audible moist rales; regular rhythm; low and dull heart sounds; and protuberant abdomen. The liver was 1 cm below the right costal margin and was soft with sharp edges. There was a palpable mass in the whole abdomen and the infant had floppy limbs and cool extremities.
The male infant was admitted to the intensive care unit for electrocardiogram (ECG) monitoring, monitoring of vital signs, blood glucose, blood gas, lactic acid and electrolytes. Multiple laboratory tests illustrated high lactic acid, high blood ammonia, high blood glucose, lactic acid and electrolytes. ECG monitoring data repeatedly verified malignant arrhythmia and he was admitted for multidisciplinary team consultation in the hospital.
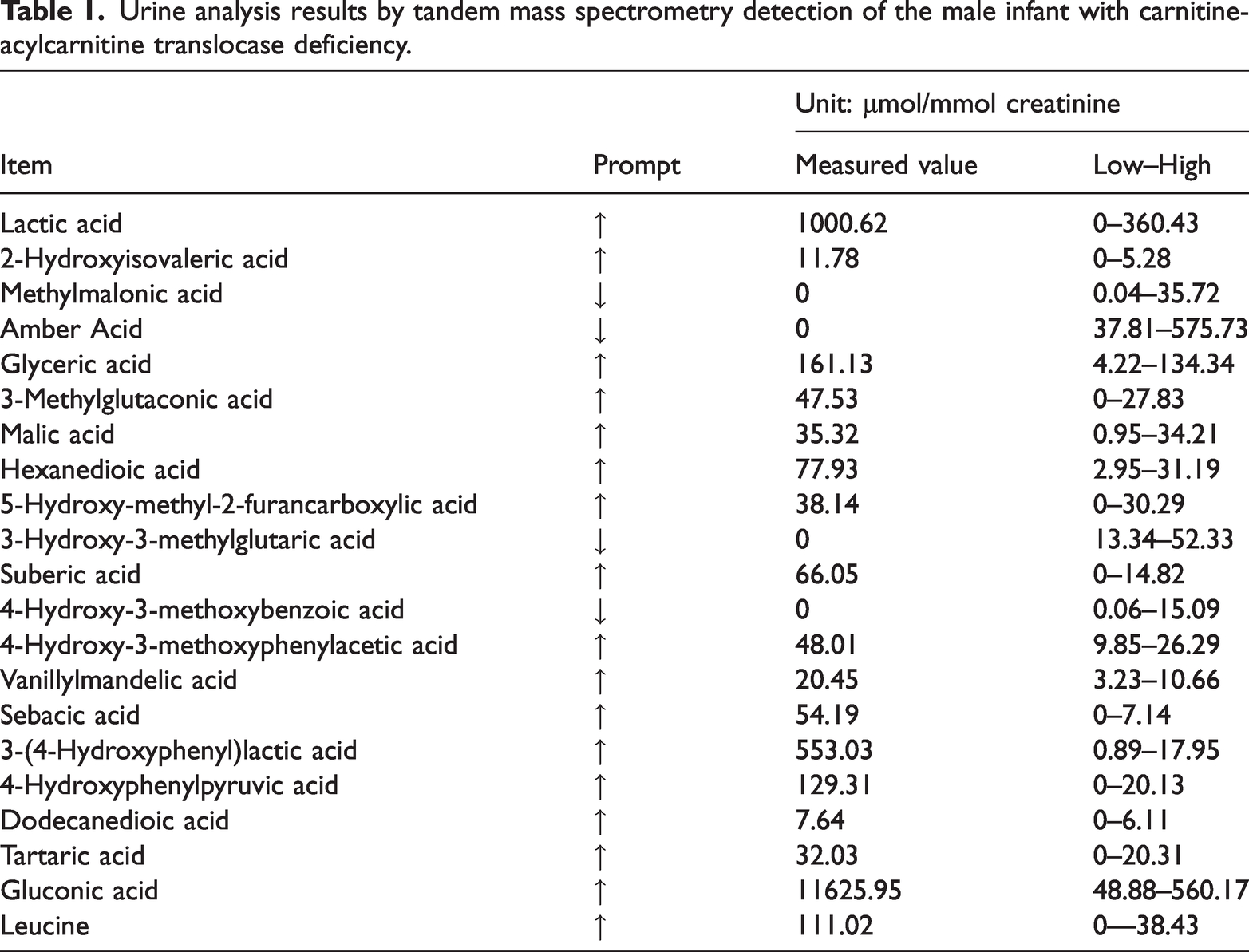
Combined with the clinical manifestations and laboratory tests, congenital metabolic diseases were highly suspected. Urine was collected from the male infant for gas chromatography-mass spectrometry analysis at the Central Laboratory of Birth Defects, Prevention and Control, Ningbo Women and Children’s Hospital, Ningbo, Zhejiang Province, China. Tandem mass spectrometry analysis demonstrated that hexane dioic acid, suberic acid and sebacic acid indicators were slightly increased in the male infant. Lactic acid indicators were increased in the male infant. 4-hydroxyphenylpyruvic acid and 4-hydroxyphenylpyruvic acid indicators were increased in the male infant (Table 1). Acylcarnitine was detected in dried blood on filter papers in the male infant (Table 2).
Urine analysis results by tandem mass spectrometry detection of the male infant with carnitine-acylcarnitine translocase deficiency.
Acylcarnitine levels of the male infant with carnitine-acylcarnitine translocase deficiency.

The male infant received the following treatment: amoxicillin and clavulanate potassium injection for infection prevention; tracheal intubation followed by ventilator to assist ventilation; multiple cardiopulmonary resuscitation; intratracheal instillation of adrenaline for pulmonary haemorrhage; adrenaline injection, dobutamine injection, dopamine injection and other vasoactive drugs for improving circulation; amiodarone injection for anti-arrhythmia; correction of acid and electrolyte disorders, abnormal blood glucose; arginine and l-carnitine for lowering blood ammonia. Electrical defibrillation was also administered, which was integrated with creatine phosphate injection for myocardial nutrition, normal saline injection, hydrocortisone injection and other symptomatic supportive treatments. The adrenaline and dobutamine injections were given intravenously to maintain the heart and improve circulation. After intravenous injection of 7 ml of 10% glucose solution, the blood glucose level was 2.1 mmol/l. After another intravenous injection of 10% glucose solution, the blood glucose level was 3.3 mmol/l and parenteral nutritional support was given at the same time. A snake venom thrombin injection was instilled into the trachea. At 04:48, his heart rate was 55 bpm and the oxygen saturation had dropped to 75%. External chest compressions were given immediately and oxygen was given by balloon pressure. After 2 min of compression, the heart rate was 58 bpm. Oxygen saturation was 70% and the infant received 1:10000 adrenaline 1 ml static push twice (at 04:50 and 04:55); and 1:10000 adrenaline 0.3 ml intratracheal instillation once (at 04:58). At the same time, continuous chest compressions combined with balloon oxygenation were given. The male infant’s heart rate rose to 110 bpm at 04:58 am and the percutaneous oxygen saturation fluctuated between 95% and 99%. Mechanical ventilation was continued and the ventilator mode was changed to high-frequency oscillation mode. The parameters were as follows: FiO2 60%; MAP 14 H2O; f 11Hz; amplitude 36 mbar. Due to a low oxygen partial pressure, the transcutaneous oxygen saturation was slightly fluctuated and the oxygen concentration increased from 50% to 60%. On 29 November 2021, the ECG monitoring illustrated ventricular tachycardia at 15:58, which was accompanied by a decrease in percutaneous oxygen saturation to approximately 80% and the rate of amiodarone was accelerated immediately from 5 µg/kg per min to 7.5 µg/kg per min. At 16:00, ECG monitoring showed ventricular fibrillation, so chest compressions were performed immediately and the oxygen concentration of the ventilator was increased to 100%. After 2 min of compression, the symptoms were not improved and the heart rate could not be measured. The transcutaneous oxygen saturation was 70% and 1:10000 adrenaline was administered (1 ml static pushing were given twice at 16:04 and 16:07) and electrical defibrillation was administered once (at 16:06). The male infant’s heart rate dropped to 52 bpm, so continuous high-quality chest compressions were given. Ventilator parameters were adjusted to the following: SIMV mode (PEEP: 6–8 cmH2O; f: 20–30 bpm; PIP: 20–22 cmH2O). The male infant was administered 1 ml adrenaline at 1:10000 once (at 16:10) and his heart rate increased to 160 bpm. At 16:12, the ECG showed paroxysmal ventricular tachycardia and 3 ml calcium gluconate was administered statically. Due to his poor response to the treatments provided, the parents withdrew treatment and he died.
After obtaining written informed consent from the parents regarding the twins, a 2-ml peripheral venous blood sample was collected from the male infant and his parents. All blood samples were sent to Shanghai Wehansi Bio-pharmaceutical Technology Company Ltd., Shanghai, China for genetic screening using high-throughput sequencing technology. Skin samples were collected from the female infant. The genetic screening mainly employed next-generation sequencing technology, containing exons and adjacent splice regions of approximately 20 000 genes (approximately 20 base pairs), as well as the detection of the total length of the mitochondrial genome. The tests were supplemented by multiplex ligation-dependent probe amplification detection, dynamic mutation detection and copy number variations analysis, which were combined with bioinformatics analysis to discover the causes of hereditary diseases and/or assess genetic risks. The pathogenicity of the variants was assessed with reference to the American College of Medical Genetics and Genomics and ClinGen Expert Group guidelines.8 High-throughput clinical exome sequencing and bioinformatics analysis of this family identified two heterozygous variants of the SLC25A20 gene in the two infants: paternal variant M1:c.706_707insT:p.R236L fs*12 and maternal variant M2:c.689C>G:p.P230R. Variant M1 resulted in a change from coding arginine to coding leucine at codon 236, followed by a termination codon in advance by frameshift. M1 has not been reported regarding CACTD previously. Variation M2 resulted in a change from coding proline to coding arginine at codon 230. Second-generation sequencing and first-generation detection demonstrated that the SLC25A20 gene variations of the male and female twin infants were inherited from their parents (Figure 1). The detection results are shown in Table 3. The first-generation sequencing results are shown in Figures 2 and 3.

Family tree showing the inheritance of two pathogenic variants of the solute carrier family 25 member 20 (SLC25A20) gene by twin male and female infants in a Chinese family. I-1, father; I-2, mother; II-2, twin brother; II-1, twin sister.
Gene variant detection results for two variants of the solute carrier family 25 member 20 (SLC25A20) gene in twin male and female infants in a Chinese family.

ACMG, American College of Medical Genetics and Genomics; OMIM®, Online Mendelian Inheritance in Man®; P, pathogenic; AR, autosomal recessive; LP, likely pathogenic.

Generation sequencing results for the paternal variant M1 of the solute carrier family 25 member 20 (SLC25A20) gene in twin male and female infants in a Chinese family. Variation SLC25A20:NM_000387.6:exon7:c.706_707insT:p.R236Lfs*12. The colour version of this figure is available at: http://imr.sagepub.com.

Generation sequencing results for the maternal variant M2 of the solute carrier family 25 member 20 (SLC25A20) gene in twin male and female infants in a Chinese family. Variation SLC25A20:NM_000387.6:exon7:c.689C>G:p.P230R. The colour version of this figure is available at: http://imr.sagepub.com.
The Ethics Committee of Ningbo Women & Children's Hospital, Ningbo, Zhejiang Province, China provided approval for publication of this case report (no. EC2021-M008). All patient details were de-identified. Written informed consent to treatment and publication were obtained from the parents of the two patients. The reporting of this study conforms to CARE guidelines. 9
Discussion
Carnitine-acylcarnitine translocase deficiency is a rare autosomal recessive inheritance that was first reported in 1993. 10 Relevant causative genes were identified in 1997. 11 The incidence rate in other countries is reported to be 0.2–1.8/100 000, 2 but the incidence in China remains unknown. Data from 1.8 million newborns that were screened in Zhejiang Province confirmed the diagnosis of CACTD in none of the patients. 12 Among approximately 150 000 newborns that were screened in Zhejiang Province, two cases were diagnosed with CACTD, which gives an incidence rate of approximately 1/76 895.12 The incidence rate in Hong Kong is approximately 1/60 000.13,14 Most of the reported cases have a poor prognosis and approximately 82% patients develop the disease in the neonatal period.4 The disease usually affects the brain, heart and skeletal muscle, mainly manifesting as convulsions, coma, arrhythmia and muscle weakness with rapid progression and high mortality.15,16 The current case report describes the clinical manifestations of twin male and female infants that were diagnosed with CACTD very early after caesarean delivery. The two infants had a very poor prognosis, did not respond well to treatment and ultimately died of the disease.
The treatment of children with CACTD includes the intravenous infusion of glucose solution during the acute phase to maximize the inhibition of lipolysis and fatty acid oxidation. Treatment during the remission phase includes avoiding prolonged fasting, limiting the intake of a long-chain fatty acid diet and supplementing medium-chain triglycerides and free carnitine. Despite these treatment options, the prognosis of most children with CACTD remains poor. Therefore, the primary and secondary prevention of this disease is particularly important. The SLC25A20 gene is located on chromosome 3p21.31 and abnormalities in the SLC25A20 gene can result in autosomal recessive carnitine-acylcarnitine translocase defects.1,17 The current case report identified two heterozygous variants in the SLC25A20 gene in the male and female twin infants; paternal variant M1:c.706_707insT:p.R236L fs*12 and maternal variant M2:c.689C>G:p.P230R. Variant M1 resulted in a change from coding arginine to coding leucine at codon 236, followed by a termination codon in advance by frameshift. Variation M2 resulted in a change from coding proline to coding arginine at codon 230. The results of second-generation sequencing and first-generation detection confirmed that the two variants were inherited from their two parents. Of these two variants, the M1 variant is a rare variant that is not included in the 1000 genome, Exome Aggregation Consortium (EXAC) and gnomAD databases. M1 has not been reported previously regarding CACTD, but it might be a candidate causal mutation for the disease. This will require further investigations to confirm. The M2 variant is a rare variant, with a frequency of 0 in the East Asian general population in the gnomAD database. The most pathogenic variants of the SLC25A20 gene are loss-of-function variants. 6 The M2 variation is a frameshift insertion mutation, which may be a non-functional variation.
Although early identification and intervention could improve the prognosis of CACTD, patients with the disease still have a high mortality rate in the first year of life. 18 The prognosis of CACTD is related to the degree of changes to enzyme activity, compliance with treatment and the rapid and effective intervention during decompensation.4,18–30
In conclusion, population genetic screening for carriers of the gene mutations associated with CACTD would be helpful for disease prevention. On the basis of a definite genetic diagnosis in parents, prenatal diagnosis and corresponding genetic counselling for the families of affected infants, as well as neonatal disease screening, would facilitate the early detection of affected patients and the timely intervention to improve their survival. Genetic counselling of affected parents is beneficial for the next healthy birth.
Footnotes
Acknowledgements
We would like to thank the four individuals that participated in this study. We want to express our gratitude to the anonymous reviewers and editor for suggestive comments, which have helped to improve this manuscript.
Author contributions
Donge Wang conceived and designed the study; Liya Zhang, Ying Hu, Min Xie, Yuxin Zhang, Kuankuan Cen, Lili Chen, Yingbo Cui, Haibo Li and Donge Wang analysed the data and drafted the manuscript.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by grants from the Medical Health Science and Technology Plan Project of Zhejiang Province (no. 2021KY324, no. 2021KY319, no. 2021KY1054); the Ningbo Clinical Research Centre for Children’s Health and Diseases (no. 2019A21002); and the Ningbo Key Discipline Paediatrics (no. 2022-B17).