
Abstract
Duchenne and Becker muscular dystrophies are the most common muscle diseases and are both currently incurable. They are caused by mutations in the dystrophin gene, which lead to the absence or reduction/truncation of the encoded protein, with progressive muscle degeneration that clinically manifests in muscle weakness, cardiac and respiratory involvement and early death. The limits of animal models to exactly reproduce human muscle disease and to predict clinically relevant treatment effects has prompted the development of more accurate in vitro skeletal muscle models. However, the challenge of effectively obtaining mature skeletal muscle cells or satellite stem cells as primary cultures has hampered the development of in vitro models. Here, we discuss the recently developed technologies that enable the differentiation of skeletal muscle from human induced pluripotent stem cells (iPSCs) of Duchenne and Becker patients. These systems recapitulate key disease features including inflammation and scarce regenerative myogenic capacity that are partially rescued by genetic and pharmacological therapies and can provide a useful platform to study and realize future therapeutic treatments. Implementation of this model also takes advantage of the developing genome editing field, which is a promising approach not only for correcting dystrophin, but also for modulating the underlying mechanisms of skeletal muscle development, regeneration and disease. These data prove the possibility of creating an accurate Duchenne and Becker in vitro model starting from iPSCs, to be used for pathogenetic studies and for drug screening to identify strategies capable of stopping or reversing muscular dystrophinopathies and other muscle diseases.
Keywords
Introduction
Duchenne (DMD) and Becker (BMD) muscular dystrophies, also known as dystrophinopathies, are the most prevalent muscle diseases, with incidences of 1:5000 and 1:18000 live born males, respectively.1–3 They are characterized by progressive weakness and muscle degeneration. Dystrophinopathies are X-linked genetic disorder caused by mutations in the DMD gene, which leads to the loss (DMD) or severe reduction/truncation (BMD) of the full length dystrophin protein.1–3 This protein is essential, both to connect the cytoskeleton with the basal lamina and to mediate signaling pathways; indeed, its absence produces membrane destabilization and subsequent muscle degeneration.4,5 Over time, the damaged fibers are not regenerated effectively and are then replaced by fat and fibrotic tissue, which causes progressive weakness with muscular atrophy and eventual death. Generally, the symptoms of DMD begin in early childhood with a rapid progression and death in early adulthood, while BMD manifests in adolescence/young adulthood and has a slower progression.
At present, there are no approved effective treatments for these diseases, because of the lack of a precise understanding of DMD/BMD pathogenesis. Currently, patients are treated with anti-inflammatory glucocorticoids, which delay disease progression, 6 drugs to treat heart symptoms, physical therapy and breathing assistance.1,7,8 Many new experimental drugs are actually under development, and some of these medications have recently been approved: ataluren permits the reading through of dystrophin nonsense mutation 9 and eteplirsen, an antisense oligonucleotide, causes the skipping of exon 51, promoting the restoration of the dystrophin reading frame. 10 Furthermore, gene and cell-based strategies are generating increasing interest.3,11–13
Animal models are essential tools in preclinical assays in order to evaluate drug effects on disease improvement and to check the consequences on other off-target tissues and behavior responses. To date, there are almost 60 different DMD animal models but in gene therapy studies DMD mouse and dog are the most frequently employed. 14 The mouse animal model (mdx mouse) is commonly used in laboratories due to its relatively low cost and accessibility, but its phenotype does not reproduce completely human muscle disease from a clinical, physiological and histological point of view. To overcome these limitations, double knockout mice for dystrophin and other muscular proteins were created in order to better mimic DMD human pathological features; however, involving a further alteration of the genetic background. On the other hand, dystrophin-deficient dogs remarkably recapitulate the human disorder clinical course and fibrotic characteristics of muscular tissue, but their use is expansive, time consuming and of low efficiency for high neonatal deaths. 14 In addition, in vivo pharmacological experiments are usually planned on homogeneous group of animals, while the next application of these treatments should be on a heterogeneous group of patients, so it is very difficult to assess the real drug effects on disease recovery. 15 As a consequence, the development of more accurate in vitro skeletal muscle models was considered to predict clinically relevant treatment effects. 3 An in vitro human skeletal muscle model can represent a useful tool for attaining a deeper understanding of muscle physiology, disease evolution, and drug efficiency or toxicity. In the past, however, the challenge of effectively obtaining mature skeletal muscle cells or satellite stem cells to serve as primary cultures has hampered the development of new in vitro models for muscular dystrophies.16,17 Furthermore, the spectrum of muscular involvement can vary, the pathological features of the disease change throughout the evolution of the disease, and these cells are not fully suitable for the analysis of all stages of this disorder or its prevention.
Recently, human induced pluripotent stem cell (iPSC) technology has allowed researchers to obtain patient-specific models of different human diseases in vitro.18–21 This technology offers an opportunity to overcome some of the issues previously faced when using a human model of DMD/BMD, allowing not only access to an unlimited number of cells, but also the potential to study the early stages of muscle differentiation. Knowledge of in vivo skeletal muscle development enabled the creation of several approaches for the differentiation of skeletal and cardiac muscle cells from iPSCs. 22
Muscle satellite cells are adult tissue-specific stem cells (muscle stem cells) found in the skeletal muscle around the muscle fibers under the basal lamina; the cellular membrane of these cells is juxtaposed with the plasma membrane of the myofiber. 23 These cells play a central role in postnatal muscle growth and regeneration in the case of myofiber damage; in fact, their ablation aggravates the atrophy in a denervated muscle fiber and prevents fiber hypertrophy in an overloaded skeletal muscle.24,25 In the case of injury, satellite cells activate the proliferation phase, a highly regulated mechanism based on the Notch signaling pathway, to produce novel myogenic progenitors that are able to differentiate and fuse for muscle repair.26,27 The new myofibers are reorganized and associated with a new group of satellite cells under the basal lamina, which is due either to their characteristic asymmetric division that allows for the formation of myogenic progenitors and the maintenance of the satellite cell pool or to their symmetric division that allows the growth of these satellite cells. 28 Unfortunately, this kind of cell is absent in cardiac muscle, limiting its regenerative capacity after injury.
The importance of cells with self-renewal and differentiation capacity is already evident during the embryonic development of muscle, when the major resident population is represented by the Pax3+/Pax7+ myogenic progenitor cells. These cells contribute both to muscle growth and to maintenance of a proliferating cell pool. In addition, during the late fetal stages these progenitor cells localize under the basal lamina occupying the satellite cell typical position and giving rise to this adult stem cell population.29–31 These cells represent a potential source for stem cell therapy approaches for DMD and BMD due to their ability to differentiate into all cell types of muscle tissue and their capacity for self-renewal throughout the lifetime of a patient. Nevertheless, the transplantation of satellite cell-derived myoblasts has not yet proven to be successful in human skeletal muscle. In fact, these cells are difficult to be expanded in vitro while preserving their staminality, and when transplanted in vivo, these cells show a limited engraftment ability and efficacy in terms of muscle regeneration. 32
Patient-specific iPSCs are expected to be a better source for in vitro disease models as well as for autologous cell transplantation therapy for DMD and BMD, because they strongly proliferate in vitro and can differentiate into many cell lineages both in vitro and in vivo. Indeed, they can provide a patient-specific disease model.
Regarding the strategies used to generate muscle stem cells from iPSCs, so far, one of the most promising methods is represented by the forced expression of myogenic transcription factors in pluripotent stem cells. For instance, the direct differentiation of human embryonic stem cells (ESCs) and iPSCs in myogenic precursors has been achieved by the conditional expression of Pax7 (a transcription factor that maintains the muscle satellite cells) and MyoD1 (a transcription factor of skeletal muscle cell differentiation and important gene in this muscle cell lineage).33–35 The first reports published demonstrated how to differentiate skeletal muscle cells from iPSC-derived fibroblasts or iPSC-derived mesodermal cells.36,37 However, new methods have been developed, excluding the redundant derivation of fibroblasts or mesoderm prior to myogenic differentiation. One of these strategies consists of the delivery of MyoD into iPSC lines by a lentiviral system. 38 This method permits the efficient production of myogenic cells, but the random integration of viral DNA can potentially cause mutagenesis events. Another possible protocol is the direct transfection of a transposon vector carrying the inducible MyoD gene,39,40 which allows for efficient and stable genomic integration. This second technique was used, for example, to differentiate myocytes from iPSCs derived from patients affected by Miyoshi myopathy iPSCs. 41
The most recently developed protocols imply the use of small molecules to direct iPSC differentiation into skeletal or cardiac muscles, thus overcoming the need for exogenous gene expression.42–44 This strategy successfully leads to the formation of robust myotubes in vitro. 45 In addition, suitable purification protocols to isolate expandable skeletal muscle progenitor cells from human iPSCs can be integrated with this strategy in order to obtain highly pure myogenic populations. 43 Using these methods Choi also produced in vivo expandable myoblasts, which can form striated contractile myofibers that participate in mouse muscle regeneration. 43
Furthermore, an important point to consider is that skeletal muscle myotubes are highly influenced by the surrounding tissue, and therefore, the substrate elasticity should mimic the native skeletal muscle environment to enhance satellite cell proliferation and to differentiate the myoblast precursors. 46 Consequently, the possibility exists of creating three-dimensional (3D) constructs in which it is possible to modulate the substrate elasticity and biopolymer coating that allow the myotubes to bind to the matrix proteins over the entire cell surface.47,48 Using this approach, it was possible to produce a 3D tissue-engineered model of DMD starting from mdx mouse primary cells, 49 and this protocol was applied to screen potential therapeutic drug candidates.
The potential to obtain two-dimensional (2D) and 3D models of muscular dystrophy is a promising element in the perspective of new therapeutic strategies development. One of these innovative technologies is gene therapy based on the microcell-mediated chromosome transfer (MMCT) of a human artificial chromosome (HAC) carrying the complete genomic dystrophin sequence. 50 This approach has already provided some significant results in iPSCs derived from DMD patients and from an animal model of DMD (mdx mouse) by allowing the correct expression of human dystrophin in muscle-like tissues. 50 Another novel strategy to correct the DMD/BMD molecular defect is the use of CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat and a CRISPR-associated 9 endonuclease system) gene editing, which has been applied in mdx mice as well as in DMD cell models with a promising correction of the phenotype.51,52 On the other hand, this new technology can be used to create iPSCs with a dystrophic phenotype, and in particular it was employed to create dystrophin-null cardiac muscle cells. 53
Studies and protocols to obtain cardiomyocytes (CMs) from stem cells are less developed than those carried out for skeletal muscle differentiation, but these systems are important since cardiomyopathy is the critical clinical feature in DMD that shortens life expectancy through rhythm disturbances, structural heart alterations and hemodynamic abnormalities. 54 For these reasons, over the last several years, a series of experiments were developed to obtain beating cardiomyocytes from the skin, blood or urine cells of patients by iPSC derivation.55–57
Overall iPSCs are expected to serve as in vitro 2D and 3D disease model systems, which will allow us to establish the pathology of muscle diseases and to develop new potential pharmaceutical approaches. However, there are still some limitations, such as the fact that a single model cannot include all of the phenotypes characterizing DMD and BMD patients. These patients have a high degree of clinical heterogeneity because there are over 1000 different mutations in the dystrophin gene that give rise to great variability in patient clinical manifestations, in addition the presence of nucleotide polymorphisms in other modifier genes can increase the discrepancy between phenotypes and the range of responsiveness to therapeutic treatments. 58 Conversely, the creation of a set of cell lines characterized by different mutations in the dystrophin gene using the CRISPR/Cas9 technology would entail an increase in costs and limit feasibility.
Therefore, in this review we summarize and discuss all of the new technologies that enable the differentiation of skeletal muscle cells from iPSCs as well as the different protocols to obtain cardiomyocytes in the context of dystrophinopathies. We explain how these strategies can provide a useful program for studying the pathological features of DMD/BMD cells, as well as the need to create a 3D model in order to better mimic the local in vivo conditions. Finally, we explore the future prospects and challenges of the therapeutic treatments under development for DMD/BMD.
Pathological features of DMD/BMD cells
Dystrophinopathies range from the most severe DMD form, characterized by precocious loss of independent ambulation, to the most heterogeneous BMD presentation, in which patients over a wide age range lose the ability to walk and present with variable cardiac and respiratory involvement. 4 This variability is due to the nature of dystrophin mutations that can give rise both to the complete loss of protein (DMD) or to the expression of partially and variably functional protein (BMD). Dystrophin is part of the dystrophin–glycoprotein complex (DGC), which comprises proteins that are essential for connecting the cytoskeleton with the basal lamina. 5 The pathogenesis of DMD is principally caused by continuous muscle contraction. In fact, the DGC absorbs the mechanical strain during muscle contraction; therefore, in the absence of functional dystrophin, the muscle fiber does not bear the high tension, leading micro breaks in the sarcolemma. 5 This situation produces a cascade of pathological effects, the first of which is the destabilization of the membrane that then becomes too permeable to extracellular calcium (Ca2+). 59 Normal muscle contraction depends on an influx of Ca2+ ions into the myotubes, but Ca2+ overload leads to mitochondrial damage and cell dysfunction or death. Over time, the damaged fibers are not regenerated effectively but are instead replaced by fat and fibrotic tissue that consequently causes muscle degeneration. In vitro muscle models have been useful in the study of these pathogenetic aspects. Through in vitro contraction experiments, excessive Ca2+ influx was demonstrated to be one of the earliest events that occurs in DMD muscle cells in response to electrical stimuli. 59 Transient receptor potential (TRP) channels expressed in muscle cells are mechanical stimuli activated Ca2+ channels through which the Ca2+ ions enter the cell.60,61 Further studies have shown that these channels can be responsible for this pathological Ca2+ influx in muscular dystrophy.62,63 The knowledge that excessive Ca2+ influx into intact myotubes under contraction is one of the primary pathological features of DMD cells and that the TRP channels contribute to DMD pathology provides a clear drug target for the treatment of DMD, which should assist scientists in finding new drug agents that can counter DMD in the early stages of disease development. 64
Other secondary pathological features identified in DMD muscle cells compared with healthy cells are the accumulation of reactive oxygen species, nuclear factor kappa beta activation, alteration of calpain activity and the release of creatine kinase (CK) into the extracellular space.65–68 Finally, other hallmarks, such as the delay in differentiation and the reduction of the proliferative capacity of satellite cells can be considered secondary effects of chronic inflammation.69–71
A recent study revealed that myoblasts obtained from the iPSCs of DMD patients and control patients, which were transfected with MyoD for differentiation, had comparable growth, underwent efficient myogenic differentiation with similar muscle specific gene expression and could efficiently fuse into physiologically and morphologically similar multinucleated myotubes with a clear striated pattern. 59 Other studies demonstrated that these myotubes were functionally responsive to treatment with hypertrophic proteins IGF-1 (hypertrophy-inducing factor insulin-like growth factor 1) and Wnt7a (wingless-type MMTV integration site family, member 7A), which are being tested as potential therapies for DMD in clinical and preclinical studies. 38 Together, these results suggest that the dystrophin alteration does not influence correct myotubular differentiation, and therefore, the patient-specific dystrophic myotubes derived from human iPSCs can be used to study in vitro the early pathological features of DMD that appear prior to the inflammatory phase. Additionally, they can be adopted as a modeling tool for new drug discovery and screening for their capacity to reproduce muscle physiology.
Differentiation protocols of iPSCs into myoblasts
A major limitation in the development of an in vitro model of muscle dystrophies as well as cell-based therapies for the cure of neuromuscular disorders is obtaining sufficient stem/progenitor cells. Because of the significant amount of muscle cells required for these types of approaches, the use of iPSCs could represent a promising resource that could eliminate this challenge. First-generation protocols for the differentiation of iPSCs into a homogenous skeletal muscle cell population were not satisfactory and usually required post-differentiation selection.72,73
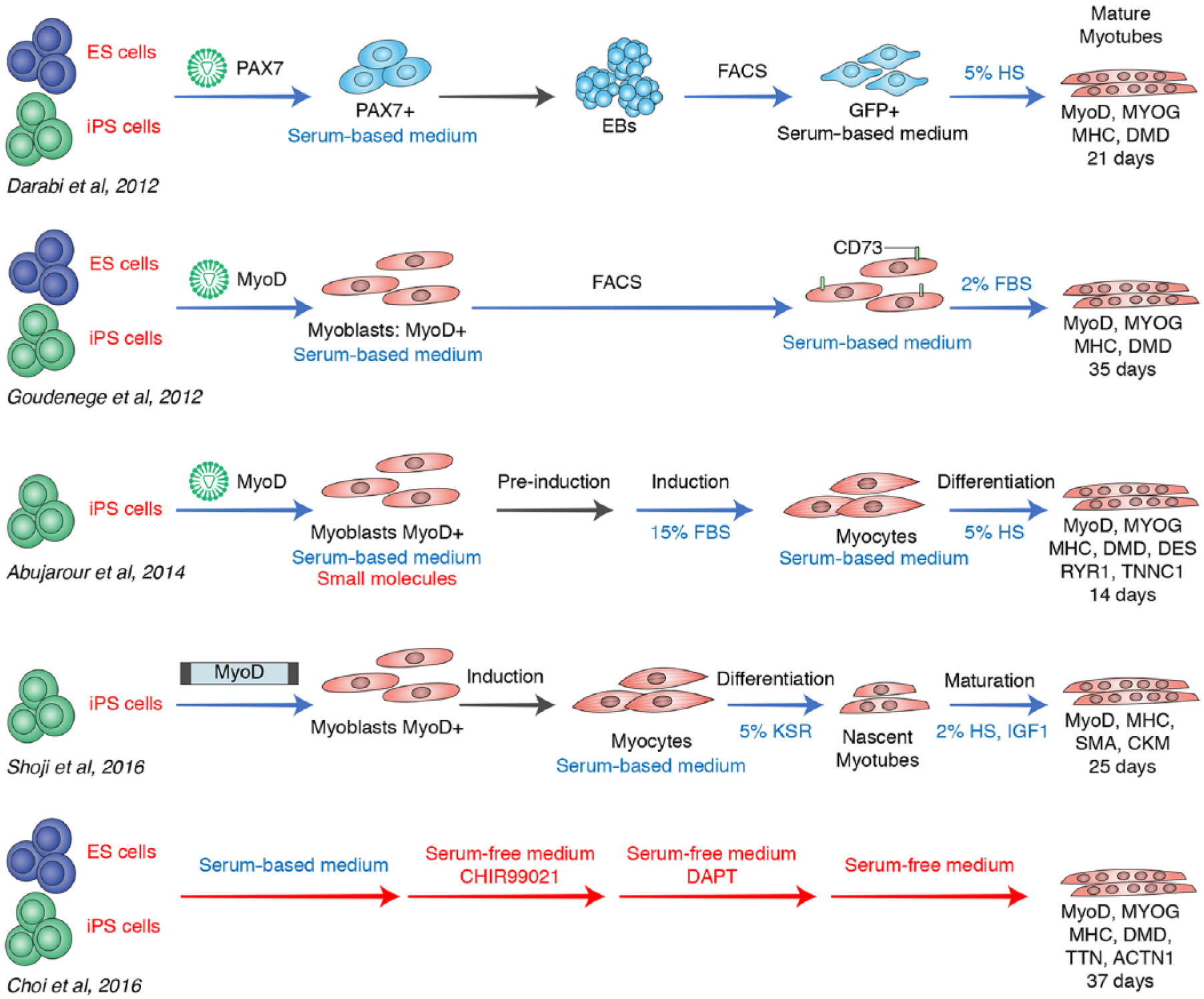
Recent protocols, however, have increased the efficiency of iPSC differentiation into myogenic cells (Table 1 and Figure 1).35,37–39,43
Main items of myotubes differentiation methods from ESCs and iPSCs.

Pre-induction medium;
Induction medium;
Differentiation medium;
Maturation medium.
2-ME, 2-mercaptoethanol; αMEM, minimum essential medium; αSARC, αsarcoglycan; ACTN1, alpha-actinin-1; bFGF, basic fibroblast growth factor; CD, cluster of differentiation; CHIR99021, inhibitor of glycogen synthase kinase 3; DAPT, inhibitor of the gamma-secretase complex; DES, desmin; DMEM, Dulbecco’s modified Eagle medium; dox, doxycycline; EB, embryoid body; ESC, embryonic stem cell; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum; HS, horse serum; IGF-1, hypertrophy-inducing factor insulin-like growth factor 1; IMDM, Iscove’s modified Dulbecco’s media; iPSC, induced pluripotent stem cell; MB1, basal medium 1; KSR, knockout serum replacement; MEF, mouse embryonic fibroblast; mTeSR, cGMP, feeder-free maintenance medium for human ESCs and iPSCs; MYL1, myosin light chain 1; N2, serum-free supplement; RYR1, ryanodine receptor 1; SMC4, small molecule cocktail 4; TNNC1, troponin C1; TTN, titin.
Advantages and disadvantages of myotube differentiation methods.
To date, most efforts directed at deriving myogenic cells from human iPSCs were based on the overexpression of myogenic transcription factors, such as Pax3, Pax7 or MyoD, by gene delivery.
Darabi and colleagues 35 reported for the first time the successful derivation of a large and proliferative population of human skeletal myogenic progenitors from both ESCs and iPSCs using conditional expression of Pax7, an important factor in the maintenance of the adult satellite cell niche.33,74,75 They genetically modified a human ESC line and two human iPSC lines with a doxycycline (dox)-inducible lentiviral vector codifying Pax7. In embryogenesis, Pax7 and its homolog Pax3 are genetic markers for the presomite mesoderm fate. Before inducing Pax7 with dox, cells were dissociated for the formation of embryoid bodies (EBs). After induction, Pax7+ cells were isolated by fluorescence-activated cell sorting (FACS) and expanded in growth medium with dox and basic fibroblast growth factor (bFGF). Both cell lines presented a great expansion capacity and were strongly positive for Pax7. They also showed a small amount of expression of myogenin and myosin heavy chain (MHC), markers of final muscle differentiation.
To induce terminal differentiation Pax7 cells were treated with a medium containing horse serum without dox and bFGF. Muscle progenitors gave rise to multinucleated myotubes with upregulation of MyoD and abundant expression of myogenin, MHC and dystrophin. These muscle progenitors were positive for CD56, which is considered a trustworthy marker of satellite cells, and they had high levels of CD63, CD146, CD105, CD90, and CD13, which are markers of mesenchymal stem cells.76,77 After transplantation into dystrophin-deficient mice, these donor cellular populations were able to efficiently create a strong engraftment and generate several dystrophin-positive myofibers. Interestingly, a little fraction of ESC and iPSC-derived Pax7+ myogenic precursors localized in the satellite cell muscular compartment, contributing to this cellular pool and they were capable of actively participating in a wide and long-term regeneration with functional recovery. 35
In the same year, Goudenege worked out a two-step protocol to obtain muscle cells from human ESCs and dystrophic iPSCs. 37 This protocol included, in the first stages, the use of a myogenic medium and, subsequently, an adenovirus infection delivering the MyoD gene. It is known that MyoD overexpression in human fibroblasts, human adult stem cells, and murine ESCs is able to initiate skeletal muscle differentiation.78–80 Therefore, the first step of this protocol was the treatment of the human ESCs with a myogenic culture medium plus fetal bovine serum (FBS), inducing a mesenchymal-like differentiation with the appearance of CD73+ cells. 72 The cells acquired a flat, spindle-like shape more similar to myogenic cells and proliferated better than myoblasts; they also lost their embryonic markers and began to produce the paraxial mesoderm markers TBX4 and TBX1. TBX4 is a key factor for the development of limb buds, 81 while TBX1 appears in the premyogenic mesoderm; 82 they are both responsible for the control of Myf5 and MyoD expression. The next step in the protocol was the infection of these cells with an adenovirus expressing MyoD and the transition to a differentiation medium. The differentiated ESCs formed numerous multinucleated myotubes in vitro and were positive for various specific myogenic markers. In fact, the authors demonstrated the presence of human myogenin and MHC, markers of terminal myogenic differentiation. 83 Once the procedure was established, the authors used this protocol on human iPSCs derived from a DMD patient to obtain myogenic cells that expressed a good percentage of MHC and fused to form large myotubes.
When transplanted into the muscle of mdx mice, both ESC and iPSC-derived myogenic cells were able to fuse with preexisting muscle fibers, according to the presence of several human nuclei positive for human dystrophin. The author also noticed a few human nuclei under the basal lamina of muscle fibers in a characteristic compartment of satellite cells, suggesting that these myogenic progenitors probably were able to form satellite cells and participate in muscle regeneration.
Despite these advances, Darabi’s and Goudenege’s approaches for the myogenic differentiation of iPSCs remain limited, since they rely on a protocol based on multiple steps. In fact, before turning on the skeletal program, there is an intermediate stage in which the EBs, mesenchymal cells, and some methods required the subsequent enrichment of differentiated cells by FACS.
Improvements in muscle differentiation protocols were made in subsequent years. Abujarour and colleagues 38 had tested the possibility of an efficient method to obtain myoblasts from disease-specific human iPSC lines, exploiting the ability of MyoD to induce direct myogenesis, 38 with the aim of drug discovery for neuromuscular diseases.78,84 This approach is based on the use of a small molecule cocktail associated with dox-inducible MyoD expression. The authors applied this protocol to iPSCs generated from healthy donors and from DMD and BMD patients. iPSCs were maintained in a growth media supplemented with the SMC4 small molecule cocktail that the authors had previously shown to have increased cell survival. Moreover, this cocktail promoted a high infection rate and a high level of MyoD expression.85,86 Differentiation was carried out in growth factor-free conditions with the addition of FBS to enhance survival and with a collagen matrix to allow attachment. Under these conditions, MyoD expression generated fast and dramatic changes in cell morphology; the iPSCs became elongated multinucleated spindle-like cells positive for MYHC. The switch from induction medium (15% FBS) to maturation medium (2% FBS) promoted the formation of striated myotubes.85,86
In addition, the characterization of myogenic differentiation by FACS demonstrated the early expression of skeletal myoblast markers, such as CD56 and CD44. To assess the possible utility of these myotubes in drug discovery, the authors evaluated their reaction to hypertrophy-inducing factors such as IGF-1 and Wnt7a. In fact, as previously described, these molecules are capable of inducing hypertrophy in skeletal muscles and can be one potential strategy for use in preclinical and clinical studies.87–89 Therefore, the treatment of the myotubes from all of the dystrophic iPSC lines with Wnt7a or IGF-1 demonstrated significant increases in diameter to levels analogous to those seen in the primary control myotubes.
These results confirm that the delivery of MyoD is an efficient approach to inducing direct myogenic differentiation that avoid the intermediate stages and FACS sorting and that the derived myotubes can potentially be used for screening drugs.
Contrary to previous studies with primary biopsied DMD myoblasts, this work demonstrated that DMD iPSC myogenic cells efficiently fused to form numerous multinucleated myotubes with a morphology similar to that of the control.90,91
These protocols efficiently produced myotubes, but they also presented some critical issues, such as the casual insertion of viral DNA, limiting cell proliferation and worsening results due to mutagenesis events, the usage of nonhuman-derived media supplements and the significant time necessary to reach a suitable endpoint.72,92
More recently, another work exploited the ability of MyoD to differentiate iPSCs directly into myocytes. 39 This protocol is based on a transposon vector carrying a dox-inducible MYOD1 that can be introduced directly into iPSCs. 41
The delivery of MYOD1 through a transposon provided uniform myogenic differentiation of iPSCs in a short period of time. In particular, this protocol included the maturation of the myotubes in a horse serum medium (2%) with the addition of IGF-1 as an enhancer. It determined the production of a homogenous multinucleated myocyte population that was positive for MHC, skeletal muscle actin (SMA) and creatinine kinase muscle type (CKM) with an efficiency of approximately 70–90%. These cells showed features of mature myocytes and were characterized by cell fusion and cell contraction when subjected to electrical stimuli. This approach was successfully used in the modeling of Miyoshi myopathy and could be applied in different kinds of patient-derived human iPSCs. In particular, it could represent a useful model for inherited diseases that require investigation of their early pathogenesis. 41
Recent protocols based on new chemical compounds, have been reported to obtain myogenic lineages starting from disease-specific human iPSCs. These approaches have been proven to be efficient at pressuring stem cells towards a myogenic pathway, leading to the robust appearance of myotubes in vitro, highlighting the fundamental role of the Wnt signaling cascade during early somite induction.42,44,45,93 Conversely, a proper purification strategy should be associated with these techniques in order to isolate expandable and very pure myogenic populations, as is generally achieved for iPSC-derived cardiomyocyte production.42,45,93
One of the current advancements, described by Choi and colleagues, 43 is a ‘chemical compound-based’ protocol able to direct iPSCs into expandable myoblasts. This strategy was applied to healthy and DMD patient iPSCs, with the aim of establish a human ‘DMD-in-a-dish’ model. 43 The protocol was developed in a few days with an initial treatment based on the use of the small molecule CHIR99021. This GSK-3b inhibitor activated the Wnt signaling pathway, supported by β-catenin translocation into the nucleus. 94 iPSCs were then cultured with DAPT, a gamma-secretase inhibitor that arrests Notch signaling and promotes myogenic differentiation. 95 Using this method, healthy iPSCs differentiated into multinucleated myotubes capable of forming striated contractile myofibers with highly organized sarcomeres. These myotubes were positive for MHC, MYOG, and MyoD, and other pivotal markers of skeletal muscle structure development and were able to actively participate in muscle regeneration in vivo.96–98 Notably, the authors also observed that a small part of transplanted human iPSC-derived myogenic progenitors expressed PAX7 and they were localized under the human basal lamina. This observation demonstrated that these cells could seed the satellite cell niche and become adult muscle stem cells, efficiently participating to the muscle regeneration.
The application of this protocol in DMD patient iPSCs led to the production of myoblasts that disclose concordant disease-related phenotypes with patient-to-patient differences, comprising abnormal expression of inflammatory or immune response genes and alterations in collagen production. These phenotypes could be partially reversed by genetic and pharmacological approaches.
It is important to notice that at the end of the treatment, both the wild-type and the DMD iPSC culture dishes displayed various types of cells, highlighting the importance of an enrichment procedure in order to obtain a purified population. The obtained myoblasts could be expanded many times, and they were easily cryopreserved without losing fusion competence. 43
This interesting protocol has demonstrated the possibility of easily generating and isolating expandable patient-specific myoblasts from disease iPSCs in a quick, effective, and determined manner. In addition, as there are some discrepancies in the same disease molecular mechanisms among DMD patients, this xeno-free model could be a useful tool with which to study these differences.
One caveat to the use of iPSC-derived skeletal muscle progenitor cells (SMPCs) was illustrated by the recent work of Hicks and colleagues. 99 They tested several differentiation protocols focusing in particular on two which are described by Chal 42 and Shelton. 93 They demonstrated that human iPSC-SMPCs obtained by directed differentiation are less functional in vitro and in vivo with respect to primary human satellite cells. They also demonstrated that human iPSC skeletal muscle is immature, but inhibition of transforming growth factor-β signaling during differentiation ameliorates fusion efficiency, ultrastructural organization and the expression of adult myosins. This maturation protocol rescued dystrophin expression in dystrophin-deficient myofibers after engraftment of CRISPR-Cas9-corrected DMD-SMPCs into the in vivo model. This study identified candidates that ameliorate the in vivo myogenic potential of human iPSC-SMPCs to levels that are comparable to directly isolated human primary fetal skeletal muscle cells. These data suggest that further studies are warranted to improve the differentiation protocols of healthy and diseased iPSCs into skeletal muscles. 99
Differentiation protocols of iPSCs into cardiomyocytes
There are two main factors motivating the efforts to obtain a model of cardiomyopathy. Cardiomyopathy is the leading cause of death in DMD patients and experimental approaches to reprogram stem cells into cardiomyocytes are far ahead of similar small molecule protocols to produce skeletal muscle.
Cardiac dysfunctions such as rhythmic disturbances, structural heart alterations and hemodynamic abnormalities develop in all patients over 18 years of age affected with DMD. 54
However, minimal data are available regarding muscle pathology and the pathways that lead to cardiac muscle degeneration. This lack of information is due both to the risk related to a patient heart biopsy as well as to the difficulty to maintain and expand cardiac cells obtained from patients in culture.
The iPSCs from DMD patients can be a source of cardiac tissue. These cells can be generated from skin biopsies, blood cells of patients and even from human urine (Table 2).53,55–57,100
Main aspects of cardiomyocyte differentiation methods from ESCs and iPSCs.

ACTN1, alpha-actinin-1; basal StemPro-34, basal serum-free medium plus nutrient supplement; bFGF, basic fibroblast growth factor; BMP4, bone morphogenetic protein 4; CTNT, cardiac troponin T; DMD, Duchenne muscular dystrophy; DMEM, Dulbecco’s modified Eagle medium; ESC, embryonic stem cell; FACS, fluorescence-activated cell sorting; IDE1, inducer of definitive endoderm 1; iPSC, induced pluripotent stem cell; IWP, inhibitor of Wnt processing; KSR, knockout serum replacement; MHC, myosin heavy chain; MLC2v, myosin light chain 2v; mTeSR, cGMP feeder-free maintenance medium for human ESCs and iPSCs; Nkx2.5, Homeobox NKX2.5; RPMI, Roswell Park Memorial Institute; VEGF-A, vascular endothelial growth factor A.
Guan and colleagues 53 generated iPSCs from progenitor cells present in the human urine of healthy controls and DMD patients and differentiated them into a monolayer culture of beating cardiomyocytes. To differentiate these cells, the authors adapted an established protocol that provided the sequential treatment with activin A and bone morphogenetic protein 4 (BMP4). 101 The iPSC colonies were detached, seeded as single cells in mTeSR1 medium and driven to differentiate by the switch in Roswell Park Memorial Institute (RPMI) medium supplemented with B27 lacking insulin and with activin A. After 24 h, activin A was exchanged with BMP4, and subsequently, this medium was replaced with RPMI-B27 plus L-glutamine medium without other exogenous growth factors. The correct temporal administration of these factors led to the production of beating cardiomyocytes in a short time. The beating cells expressed cardiac markers such as sarcomeric α-actinin, cardiac α and β MHC, as well as membrane-localized connexin-43. Additionally, quantitative analysis demonstrated the upregulation of cardiac genes. The functionality of these differentiated cardiomyocytes was tested by subjecting them to patch clamp recording and displaying the presence of spontaneous action potentials (APs). 53
Notable differences between DMD and normal cardiomyocytes were evidenced. In particular, only cardiomyocytes from wild-type controls were positive for dystrophin, while the DMD cardiomyocytes preserved their dystrophin-deficient phenotype. In addition, divergences were noted in calcium uptake: the length of recovery of calcium in DMD patients was extended compared with the controls, demonstrating, therefore, that the mitochondrial permeability pore opening was altered. Finally, the hypotonic stress analysis showed that the cardiac injury markers CK-MB and cardiac troponin I (cTnI) were higher in DMD than in normal cardiomyocytes.
In conclusion, cardiomyocytes generated from the urine of DMD patients preserved the dystrophin-deficient phenotype and showed distinctive characteristics that might be further harnessed in mechanistic studies. Moreover, physiological assays supported the feasibility of using urine-derived cardiomyocytes as a biological reagent to find new compounds or test existing drugs, which could protect dystrophin-deficient cardiac cells from stress-induced damage.
In another interesting study, Lin and colleagues 55 developed an in vitro model to replicate the features of dilated cardiomyopathy of DMD patients in order to study the underlying disease mechanism. 55 In this study, human control and DMD dermal fibroblast-derived iPSCs were differentiated in cardiomyocytes by the formation of EBs. The EBs were treated with BMP4, bFGF and activin A in order to activate the Wnt signaling pathway. After a few days to allow for the enhancement of the cardiac induction, the authors repressed the Wnt/β-catenin pathway using XAV-939, a Wnt inhibitor associated with VEGF (vascular endothelial growth factor). After 22 days of differentiation, they obtained EBs exhibiting spontaneous contractions. The iPSC-cardiomyocytes (iPSC-CMs) that resulted from the dissociation of the EBs were positive for anti-cardiac troponin T (CTNT). Moreover, DMD iPSC-CMs showed decreased levels of dystrophin and presented enhanced levels of cytosolic Ca2+, mitochondria damage, caspase-3 activation and cell apoptosis compared with the control CMs. The authors also performed transcriptional and translational analyses, which found that the augmented death in DMD iPSC-CMs could be caused by an altered mitochondria-mediated signaling pathway, and that this harmful network could be pharmacologically modulated.
A more recent paper described the successful differentiation of DMD patient iPSCs obtained from T lymphocytes into cardiomyocytes and demonstrated that these cells could be a useful in vitro experimental system. 56 The iPSC lines were directed towards the cardiac fate using an EB formation approach. The iPSC colonies were detached and plated in small clusters in low attachment dishes with a feeder-free culture medium plus growth factors. 56 To induce differentiation, the cells were treated with CHIR99021, IDE1 (inducer of definitive endoderm 1) and bFGF, and subsequently, the Wnt pathway was inhibited with IWP-2 (inhibitor of Wnt processing), VEGF, SB431542 (a potent and selective inhibitor of activin A receptor-like kinase ALK5), and dorsomorphin. Finally, the differentiated cardiomyocytes were purified by metabolic selection using a glucose-free medium with lactate. Following this differentiation/purification method, spontaneously beating EBs were expeditiously obtained in different experiments. Cardiac-specific markers, including alpha-sarcomeric actinin and myosin light chain 2v (MLC2v) were abundantly present in cells gained from beating EBs from the control and DMD patients; however, the DMD-EBs lacked dystrophin expression, while this protein was highly present in the controls. The data suggested that the cells obtained from beating EBs showed the general properties of cardiomyocytes, and the DMD beating EBs mirrored the patient phenotype. While these results were interesting, only a few cell lines were obtained and analyzed therefore, many experiments should be performed to make this protocol reproducible.
All these approaches have increased our knowledge of the molecular pathways involved in the genesis of cardiomyocytes starting from stem cells, but in order to be useful, the application of in vitro cardiac tissue models should comprehensively reproduce disease stratification states in healthy samples to develop novel approaches to rescue the disease phenotype.
3D models and organoids
In the last few years, some 3D skeletal muscle models have been developed in order to study the molecular phenotypes of neuromuscular disorders (Table 3).102–107
Main materials and assays to generate 3D muscular models.

3D, three-dimensional; iPSC, induced pluripotent stem cell.
While different assays demonstrated the ability of neural stem cells to self-organize in 3D structures resembling whole organs, 108 the equivalent 3D model for muscle is still evolving to attain major complexity. To develop genetic engineering approaches, high amount and high-throughput cellular in vitro assays are required. For this purpose, in the past, the immortalized murine myogenic cell line C2C12 had been an important source, and it was used in engineering the skeletal muscle tissue due to its availability, cost effectiveness and reduced variability. 109 More recently, ESC and iPSC-based models have been shown to be able to develop skeletal muscle cells with high efficiency, 110 but unfortunately, they lack environmental factors, regional identities and architectural scaffolds compared with the tissue samples of animal models or human patients. In addition, highly faithful 3D models require the presence of satellite cells able to proliferate in order to maintain a quiescent satellite cell population and differentiate into functional skeletal muscle. 111
In general, to generate 3D models, myoblasts should be seeded into 3D matrices and treated with a differentiation medium that induces myoblasts to fuse into myotubes. These cells are able to align in the structure through adhesion points, respond to mechanical cues and reorganize themselves within the scaffold. Matrigel, collagen, fibrin, or synthetic polymers such as lactide-co-glycolide, and caprolactone are scaffold-tested for such muscle constructs. It is unclear how different matrices contribute to reproducing the muscle architecture influencing myogenic activity in culture. 112 Penton and colleagues 102 tested different extracellular matrices such as Matrigel and some laminins in order to evaluate cell differentiation capacity over long-term expansion. While Matrigel performed strongly in short-term mouse studies, in long-term expansion, it demonstrated an excessive rate of variability in differentiation and could not be used for clinical applications. Furthermore, these authors demonstrated that laminin 211 (constituted by α2, β1, λ1 chains), the native isoform in resting skeletal muscle, resulted in low proliferation and poor differentiation in both mouse and human cultures. Instead, laminin 521 (constituted by α5, β2, λ1 chains) was a safe substrate to expand myoblasts for cellular therapy in clinical studies because it was a valuable matrix for short and long-term muscular cell maintenance; its performance was superior to other types of laminins, gelatin and Matrigel. The observed results regarding laminin 521 were probably due to the presence on the α5 chain of a higher number of binding sites for integrin α3, αV, and α6β4 that are not present in other laminin isoforms (Table 3). 102
Other strategies used to ameliorate the 3D models and to induce the generation of directed forces in bioartificial muscles are co-cultures with motor neurons and the treatment with broad-field electrical stimulations. Morimoto and colleagues 103 used neurospheres derived from mouse neural stem cells, kept in contact with the skeletal muscle fiber bundles. They demonstrated that these neurospheres were able to differentiate into neurons and to drive the formation of neuromuscular junctions (NMJs). When the neurons were activated by glutamic acid, there was an acetylcholine release. The interaction of this neurotransmitter with the acetylcholine receptors localized on NMJs enhanced a unidirectional contraction in the neuron–muscle constructs. These results were validated by the treatment with curare, an NMJ antagonist, which blocked the contraction (Table 3). 103
Conversely, Demestre and colleagues 104 generated co-cultures of motoneurons and myotubes derived from the same human iPSC cell line. The myotubes displayed the early aggregation of acetylcholine receptors and, upon electrical stimulation, showed a contraction as a consequence of neuronal acetylcholine release (Table 3). 104 These co-cultures were stained with Bassoon, a presynaptic marker that tags axon synaptic contacts with a neurofilament (NF) marker that recognizes neuronal axons, and with α-bungarotoxin, which marks the NMJs. In this way the authors detected NF-positive axons close to α-bungarotoxin-labeled NMJs, demonstrating the presence of the endplate areas, together with the Bassoon staining of small punctae, indicating pre- and post-synaptic contacts. The advantage of this protocol was the creation of a functional system consisting of two different communicating cellular types derived from the same human iPSC line; this approach, therefore, could open the way to future patient-specific experimental therapies.
The most significant challenges regard the development of fully matured skeletal muscle fibers to adequately model adult physiological architecture. Juhas and colleagues 105 assessed a 3D in vitro environment primed for myogenic maturation that consisted of activated rat Pax7+/MyoD+ myogenic cells, which underwent rapid fusion and the formation of aligned myofibers. Subsequently, the newly formed muscle bundles anchored at the ends to polydimethylsiloxane wells that provided the needed tension for final maturation. Such skeletal muscle constructs developed native-like niches containing quiescent satellite cells and a basal lamina-like matrix formed by laminin and collagen IV. These 3D structures, implanted dorsally in a mouse model, were able to produce vascularization and perfusion, satellite cell niches maintenance and sustained myogenesis. To evaluate calcium handling and contractile function, the primary myogenic cells of the construct were transduced with a fluorescent intracellular calcium indicator, showing improvements in these parameters and force levels comparable to those of native tissue (Table 3). 105 Recently, the same group applied a similar efficient protocol, starting from human iPSCs, to produce Pax7+-induced myogenic progenitor cells that underwent a rapid differentiation if embedded in fibrin-based hydrogels and cultured in media for 3D tissues. Similar to the previous report in rats, the obtained human muscle bundles contained aligned cross-striated myotubes and Pax7+ satellite cell pools. These skeletal muscle constructs could contract if electrically or chemically stimulated and showed vascularization and perfusion if implanted in immunocompromised mice (Table 3). 106 In the same year, Maffioletti and colleagues 107 obtained the first 3D skeletal muscle model based on human iPSCs derived from patients with different muscular disorders such as limb girdle 2D muscular dystrophy, skeletal muscle laminopathies, and in particular from DMD. The myogenic differentiation was reached using biocompatible fibrin-based hydrogels under tension to induce myofiber alignment. These patient-specific artificial muscles showed the pathological hallmarks of severe muscular dystrophies. To mimic the in vitro skeletal muscle complexity characterized also by the presence of different nonmuscle cell types supporting the vascularization, perfusion and innervation, the authors derived different human iPSC isogenic lineages, including vascular endothelial cells, pericytes and motoneurons, creating the first four lineage isogenic system (Table 3). 107
Adaptation of these systems will be of fundamental importance for the production of reliable skeletal tissue models for high volume drug screening testing. 113
3D cardiac tissue
Pluripotent stem cell-derived cardiomyocytes (CMs) are more similar to fetal CMs than to mature CMs, and this feature drove their application in cellular therapies and drug screening. As cell behavior and maturation are influenced by extracellular matrix and 3D structures, different materials and assays have been developed (Table 4).114–119
Materials and assays to generate 3D cardiac models.

3D, three-dimensional; GTPase, guanosine triphosphatase; iPSC, induced pluripotent stem cell.
Fong and colleagues 114 demonstrated that the final differentiation of iPSC-derived CMs was boosted if the cells were spread onto 3D cardiac scaffolds. This treatment enhanced the expression of Junctin, CaV1.2, NCX1, HCN4, SERCA2a, Triadin, and CASQ2, which are implicated in calcium handling and cardiac contraction. 114
Actual methods for engineering cardiac tissue on scaffolds require endothelial cells, smooth muscle cells, myofibroblasts, mesenchymal stem cells and, in particular, cardiac fibroblasts that are important for contractile force improvement, producing angiogenic factors essential for a stable formation of endothelial cell sprouts and subsequent vessel maturation in vitro. As demonstrated by Twardowski and Black, 115 endothelial cells developed more sprouts when these cells were co-cultured with cardiac fibroblasts in 3D fibrin hydrogels; on the contrary, co-culture with mesenchymal stem cells induced multicellular sprouts, suggesting that these cells were able to enhance the adhesion and protrusion. 115 These cells are supposed to recreate the correct radial and longitudinal pulse stresses, 120 mimicking the tissue architecture, structural properties and electromechanical coupling of a native cardiac extracellular matrix.
Therefore, Kim and his colleagues 116 reported their findings on in vitro modeling of autologous cardiosphere-derived cells seeded in the nanotopographical polymeric substrata of a hydrogel. The treatment of these cardiospheres with a GTPase activation protein for the small G-protein RhoA, mediated the transduction of substrata extracellular mechanical signaling into a cellular transcriptional response, inducing the cardiomyogenic differentiation of cells. Engraftment of these scaffolds in a rat infarction model enhanced the retention and growth of the transplanted cells and their integration with the host tissue, reproducing structural and functional cardiac properties. 116
To date, however, the engineered cardiac tissue for high-throughput drug screening assays is not fully developed; the major hindrance to the application of these technologies is the inability to test parameters such as cell source variability, mechanical, soluble and electrical stimuli. Boudou and co-workers 117 reported a new method to produce mini-engineered heart tissue, formed by cardiac cells embedded in collagen and fibrin 3D matrices, grown into microgauges; these nanotechnologies were able to check and record the cardiac microtissue-generated forces in real time. This microtissue assay was tested with isoproterenol and digoxin and demonstrated reproducible, dose-dependent effects on contractility and beating frequency. 117 Nevertheless, in situ real-time monitoring is a critical step to exactly estimate the reaction of cardiac tissue to experimental drugs. Therefore, Zhang and colleagues 121 created a pillar electrode platform inserted into microchannels for growing and electrically stimulating 3D cardiac tissues in order to assess the dynamic behavior of these microtissues in situ over a period of weeks. 121
The correspondence of structural and functional characteristics between these assays and in vivo heart muscle is promising and opens the potential to high-throughput, low-volume drug screening and future applications.
The first positive results obtained by these nanotechnologies opened the door to new approaches in order to study DMD patient cardiac features.118,119
Macadangdang and colleagues 118 highlighted the DMD-altered specific features relating to cell structure and contractile function by means of an easy biomimetic engineered platform able to enhance the development of cardiomyocyte structure and morphology. These anisotropic nanofabricated substrata (ANFS) were composed of parallel groups of channels and chines, simulating the methodical structure of the myocardial extracellular matrix. In this study, human iPSCs from healthy controls and a DMD patient were produced from isolated urine cells and subsequently induced to differentiate into a monolayer cardiac lineage. After induction, the iPSC-CMs were detached and cultured flat or on ANFS to compare the influence of these two different environments on cellular behavior. With regard to the subcellular structure, normal iPSC-CMs displayed a greater level of anisotropy and alignment on the ANFS compared with the flat control. In particular, the actin cytoskeleton showed local alignment in the flat condition without preferential directionality, while on ANFS, it was highly aligned in the major direction of this nanostructure. Even DMD iPSC-CMs on flat substrates were casually arranged as usual but presented lower directionality than normal cardiomyocytes on ANFS. Therefore, these results indicated that healthy human iPSC-CMs aligned with the underlying anisotropic nanotopographic sticks, while DMD iPSC-CMs demonstrated the lowest macroscopic alignment and a decreased capacity to respond to topographic cues compared with normal iPSC-CMs. 118
Since in clinical settings, the cardiac involvement in DMD is commonly related to cardiomyopathy complicated by arrhythmias, 122 the authors decided to measure the cell area to determine if this phenotype was present in their DMD cardiomyopathy model. In DMD iPSC-CMs the cell areas were significantly greater than normal, and the resting sarcomere lengths were remarkably longer than normal ones. These two findings illustrated that DMD iPSC-CMs seemed to retain some of the main structural traits of DMD cardiomyopathy and suggested a definite model of ‘dilated cardiomyopathy in a dish’.
The authors also performed contraction analyses in which they observed that DMD iPSC-CMs showed quicker contraction velocities than normal iPSC-CMs. A lack of dystrophin is known to disrupt the link between dystrophin and the glycoprotein complex (DGC), leading to a higher vulnerability of the myocytes to mechanical stress, without any alteration in the cardiac differentiation. 123 Therefore, the authors hypothesized that the major contraction and relaxation kinetics observed in DMD cardiomyocytes could be due to a weak attachment to the underlying substrate and, consequently, the DMD cardiomyocytes were more capable of contracting than the controls, leading to an apparent gain of function compared with normal cells.
Overall, the different behavior of the diseased and normal cardiomyocytes to ANFS described by the authors is indicative of a structural and functional cardiomyopathy-like phenotype in DMD human iPSC-CMs. Therefore, the reported engineered system could represent better DMD model of cardiomyopathy in vitro to use in drug screening assays and studies on muscular mechanics. 118
In a more recent study, Carson and colleagues 119 applied a similar approach based on the use of a nano-grid culture array formed by nano-grooved topographies similar to the complex extracellular matrix structure of the myocardial basement membrane. These substrates were similar to fibronectin-treated surfaces since they were linked to a chimeric peptide characterized by the presence of a cell adhesion motif (Arg-Gly-Asp) that enhanced the cell matrix binding. The protocol began with iPSC colonies derived from fetal lung fibroblasts, 124 which were seeded into a single cell monolayer to enhance differentiation. The mature cardiomyocytes were dispersed and re-plated on experimental nanopatterned surfaces of different scale. This method allowed for the analysis of the consequence of some nanoscale structures on human iPSC-CMs differentiation in vitro, and in fact, the organization and structural evolution were influenced by the nanotopographical material size. 119 The authors highlighted that cardiomyocytes cultured on specific grooves (700–1000 nm range) were able to orient into a parallel order that more closely mimicked the native mammalian myocardium structure.
Therapeutic perspectives
In recent years, several therapeutic approaches aimed at modifying the natural course of DMD have been tested in both in vitro studies and clinical trials (www.clinicaltrials.gov). They include drugs that act to restore dystrophin expression, and molecules, that act on the pathogenic cascade determined by the absence of dystrophin. Some of these drugs have even been introduced into clinical practice. In fact, recently TranslarnaTM (ataluren), a read-through agent, 125 has received conditional approval in the European Union as treatment of ambulatory DMD patients with nonsense mutations and eteplirsen, an antisense oligonucleotide that promotes exon 51 skipping, has also been approved for patients carrying the suitable deletions. 126 Unfortunately, these new and promising compounds have limited efficacy when the signs and symptoms of the disease are already present. Therefore, muscular dystrophies remain incurable disorders, and the creation of new models with which to study new potential therapeutic approaches is essential.
iPSCs could be used not only to better understand the pathogenesis of the disease, but also to establish a valid asset for evaluating the efficacy of new therapies specifically developed for common forms of muscular dystrophies. Mature skeletal myocytes or satellite cells can be obtained only with invasive procedures and cannot be expanded indefinitely in culture systems. In contrast, iPSCs can be generated from different tissues and represent an unlimited source of self-renewable cells able to differentiate into any cell type, including myoblasts. 38 Thus far, the majority of preclinical trials have been performed on animal models such as the mdx mouse; however, these models present phenotypical and physiological differences compared with human disease. For example, in the mdx mouse model, the clinical phenotype is milder than in DMD patients, with a late onset of cardiomyopathy and fibrosis. 127 This difference could partially be to blame for the failure of some clinical trials. In fact, when translated into human clinical trials, many drugs investigated using the mdx model showed previously unreported collateral effects and lack of efficacy. 128
The knowledge of the genetic defect responsible for DMD makes gene therapy a valid tool for the treatment of the disease. So far, several techniques had been used to improve muscular function and restore dystrophin levels in these patients; some of these techniques have also been evaluated using iPSCs.
Gene therapy
An approach for correcting dystrophic phenotypes is represented by the incorporation of dystrophin, one of the largest human genes (2.4 Mb), into the cells.
Since viral vectors, such as adeno-associated virus (AAV) or lentivirus, are able to contain only limited amounts of genetic material, shorter versions of the DMD gene were created in order to produce a smaller but still functional protein, shifting the phenotype from DMD to BMD-like. Mini-dystrophins contain only essential domains, generally without regulatory sequences. Despite the reduced packing capacity in viral vectors, it is important to make an effort to include critical functional motifs, such as the neural nitric oxide synthase (nNOS) binding site, in order to enhance therapeutic efficacy. The importance of these particular domains was demonstrated by Lai and colleagues (2009) 129 with AAV transfection experiments of deleted dystrophin constructs in mdx mice. The authors demonstrated that the presence of spectrin-like repeats 16 and 17 in the protein was essential for nNOS anchoring to sarcolemma. The mice, treated with synthetic dystrophins containing these binding sites, improved their muscle pathology, muscle strength and exercise performance. 129 More recently, Meng (2016) 130 applied this strategy lentivirally transducing the vector into muscle stem cells (pericytes) of a DMD patient, and these cells, when transplanted into a mdx nude mouse, regenerated muscle fibers and restored functional dystrophin expression and nNOS function. 130
Analogue techniques could be tested on human iPSCs to further evaluate the unsolved issues of viral-mediated approaches, such as the risk of insertional mutagenesis. For instance, Zhao and colleagues 131 developed a plasmid-mediated method, avoiding random genome integration. A full length dystrophin cDNA (⁓14 kb) was inserted using Bxb1 integrase into a precise location of the mdx mouse iPSC genome. These cells were able to differentiate into myogenic precursors in vitro, and when they were transplanted into the mdx mouse muscle, they engrafted and originated myofibers expressing dystrophin. 131 To improve the efficacy of gene therapy and to reduce the risk for insertional mutagenesis, HACs were created. They are exogenous mini chromosomes that stably remain episomal in the host genome, replicate and segregate during mitosis and carry large genetic loads. Therefore, this kind of structure can contain the entire genomic dystrophin (DYS-HAC), including the regulatory elements.132,133 DYS-HAC could correct iPSCs derived from DMD patients (carrying exon 4–43 deletion) or treat mdx mice if transferred into fibroblasts via MMCT. Even after 4 months, HAC was stable in iPSCs, derived from these transfected fibroblasts, as an individual nonintegrating chromosome; those iPSCs were able to differentiate in embryonic germ layers and expressed human dystrophin in muscle-like tissues of teratomas. Notably, it was not possible in this study to directly transfer DYS-HAC into iPSCs due to the difficulties in cloning colonies derived from single cells after transfection or MMCT. 50 Later it was also proven that DYS-HAC could be transferred through MMCT into mouse ESCs and mesangioblasts isolated from mdx mice with amelioration of the phenotype. 134
Exon skipping
Antisense oligonucleotides (AONs) are small chemically modified nucleic acids designed to target specific gene transcripts, and induce the skipping of specific exons during mRNA splicing to restore the reading frame of selected out-of-frame mutations, thereby leading to the creation of a smaller but still functional proteins.135,136 Antisense oligonucleotide AO88 skips exon 45 in human iPSCs derived from patients carrying a deletion in exon 44 and 46–47, restoring dystrophin expression and reducing calcium overflow and secretion of CK in DMD myotubes. 59 To date, some clinical trials have been developed to evaluate AONs to promote the skipping of exon 51, 44, 45 and 53 and in the near future, the effects of customized AONs specifically created for different deletions can be assessed using iPSC models. 137
Genetic engineering
Another tool for genetic correction of iPSCs is represented by genome editing, using the transcription activator-like effector nuclease (TALEN) or, more recently, the CRISPR and Cas9 endonuclease system. These nucleases allow precise genome editing in cultured cells, inducing site-specific DNA cleavage. 138 However, the safety in terms of mutagenesis of this approach is still to be determined.
Li and colleagues 139 compared three different methods for genetic correction of iPSCs derived from a DMD patient with exon 44 deletion using nucleases: the disruption of the splicing acceptor of exon 45 in order to restore the reading frame, the introduction of small indels to induce a frameshift, and the knock-in of exon 44 to restore the full protein coding region. All these approaches restored dystrophin expression in differentiated skeletal muscle cells, even though exon knock-in seemed to be more effective than exon skipping and frameshift. 139
Genetic modification can also be obtained through the Sleeping Beauty transposon system, a synthetic transposon capable of introducing specific DNA sequences into chromosomes. 140 Transplantation of myogenic progenitors derived from dystrophic iPSCs corrected with Sleeping Beauty transposon carrying the micro-utrophin gene in dystrophin-utrophin double knockout mice restored the DGC and improved contractile strength. 141 In this case, these mice were used, since utrophin is a dystrophin-related protein normally upregulated in the sarcolemma of mdx mice and therefore mdx-utrophin mutants showed a severe phenotype in comparison with mdx mice.
Stem cell transplantation
Another therapeutic strategy consists of the transplantation of muscle stem cells, derived from patients or from healthy controls, that are administered locally or arterially. A phase I–IIa clinical trial with intra-arterial human mesangioblast transplantation was performed in DMD patients. Unfortunately, no functional improvements were observed, 142 nevertheless mesangioblasts seemed to ameliorate the dystrophic phenotype in mouse and dog models. 143 However, this approach required a significant number of cells, and mesangioblasts have a limited life span. iPSC-derived mesangioblasts (HIDEMs) could represent a possibility, providing an unlimited source of cells. This method was used in limb girdle muscular dystrophy 2D, which is due to a mutation in the gene encoding α-sarcoglycan (SGCA gene). HIDEMs were generated from iPSCs and were corrected with cDNA encoding SGCA and MyoD. Intramuscular administration of these cells in SGCA-null mice demonstrated muscle regeneration, which was also demonstrated when mice were injected via the arterial route.144,145 Because the immune response against integrated viral vectors, against the allogenic mitochondrial genome and even against syngeneic mouse iPSCs has been reported, the authors also studied the immunogenicity of these cells; HIDEMs not only had similar myogenic potential to that of normal mesangioblasts, but also equivalent immunomodulatory functions, exerting immunosuppressive effects on T-cell proliferation. 146
Always trying to avoid an immune response triggered by allogenic antigens, another source of cells for replacement therapies may be represented by genetically engineered corrected autologous iPSCs. Human iPSCs cultured in a myogenic medium and infected with an adenovirus expressing MyoD may be differentiated into cells able to participate in muscle regeneration by fusing with the remaining muscular fibers when transplanted into Rag/mdx mice. 37
Therapeutic perspectives in cardiomyocytes
Cardiomyopathy is an important aspect of the DMD phenotype, and after the advent of nocturnal ventilation and steroid treatment, heart failure has emerged as a leading cause of death in these patients. 147 Cardiomyocytes differ from skeletal muscle cells in terms of both dystrophin mRNA levels and turnover. Furthermore, the half-life of drugs is longer, and delivery seems to be less efficient than in the skeletal cells. 148 Therefore, it is important to develop reliable in vitro models in order to test the effects of different drugs specifically on the cardiac muscle. Moreover, primary human cardiomyocytes cannot be obtained easily or divide in culture, and as a consequence, different approaches have also been tested in human iPSC-derived cardiomyocytes.
Dick and colleagues analyzed the exon-skipping efficacy in cardiomyocytes derived from human iPSC originating from skin fibroblasts. 149 Antisense oligonucleotide directed to exon 51 was transfected into iPSC-CMs obtained from DMD patients carrying frame-shifting deletions (deletion 47–50 or 48–50) or nonsense point mutations leading to the expression of truncated RNA transcripts. Immunostaining performed 3 days after transfection revealed increased dystrophin protein levels (30% more than wild-type cells).
However, exon skipping is not suitable for every type of mutation. To overcome this issue, mini dystrophin delivery was also tested in iPSC-CM cells. Mini dystrophin constructs of 4.5 kb were lentivirally-delivered to DMD human iPSC-CMs carrying a stop mutation in exon 70 or a dinucleotide deletion in exon 35, inducing dystrophin minigene expression at levels of 66–91% of healthy control cardiomyocytes, with a transduction efficiency of 55%. 149
Human cardiomyocytes derived from DMD-iPSCs and corrected with DYS-HAC maintained DYS-HAC during differentiation to EBs and displayed activation of multiple promoters of the dystrophin gene, expressing different dystrophin isoforms and correctly localizing to the membrane. 150 By capitalizing on CRISPR-technology and focusing particularly on the match between the eukaryotic splice acceptor and splice donor sequences and the protospacer adjacent motif sequences that govern prokaryotic CRISPR/Cas9 target gene recognition and cleavage, Long and colleagues 151 carried out editing in iPSCs from different DMD patients with large deletions, point mutations, or duplications and efficiently rescued dystrophin protein expression in cardiomyocytes. In 3D heart muscle systems, correction of DMD mutations rescued dystrophin expression and the mechanical force of contraction. Editing only a fraction of cardiomyocytes (30–50%) was enough to improve the mutant phenotype to near-normal wild-type levels. The author demonstrated that eliminating conserved RNA splicing acceptor/donor sites and addressing the splicing machinery to skip mutant or out-of-frame exons through editing permitted correction of the cardiac abnormalities associated with DMD. 151
Moreover, studies about the underlying pathogenic mechanisms of DMD cardiomyopathy using iPSCs demonstrated that treatment with the membrane sealant poloxamer 188 reduced pathological aspects such as resting cytosolic calcium levels, caspase-3 activation and apoptosis. 55
Regarding cell transplantation studies, it has been shown that wild-type mouse iPSCs injected into mdx mouse and mdx-utrophin mutant blastocysts corrected the morphological and functional features of mdx skeletal muscle and normalized the fat–body weight ratio in both models. 152 Furthermore, Pax7-induced iPSCs gave rise to a proliferating population of myogenic progenitors that promoted substantial muscle regeneration and contributed to the improvement of the contractile properties when injected intramuscularly into dystrophic/mdx mice; regeneration and engraftment of donor cells were facilitated using immunosuppression and preinjury with cardiotoxin. 35
Finally, using human iPSCs for drug development enabled the possibility of making extensive in vitro assessments of multiple parameters in order to evaluate toxicity and efficacy in a human model. To date, pharmacological responses of nondystrophic human iPSC-CMs to over 100 compounds have been demonstrated. 153
Current limitations and future perspectives
In recent years, several therapeutic approaches to modify natural history of DMD have been tested in both in vitro studies and clinical trials, from drugs favoring the restoration of dystrophin expression to molecules acting on the pathogenic cascade.125,126 Unfortunately, these new and promising compounds have not been able to stop muscular degeneration but can only slow disease progression. Therefore, muscular dystrophies remain incurable disorders, and the creation of new models in which to study potential therapeutic approaches is essential.
A major limitation in the development of in vitro models of muscle dystrophies as well as cell-based therapies for the cure of neuromuscular disorders is obtaining a sufficient number of stem/progenitor cells for their application. Moreover, the spectrum of muscular involvement of DMD and BMD can vary widely, and the pathological features change during disease evolution. Recently, the use of patient-specific human iPSCs offers an opportunity to overcome some of these issues because these cells are more suitable for the analysis of the early stages of muscle differentiation of these disorders and their preventive measures. In addition, patient-specific iPSCs are expected to be a better source, even for autologous cell transplantation therapy for DMD and BMD, avoiding immune reactions because they strongly proliferate in vitro, and they can differentiate into many cell lineages both in vitro and in vivo. An important assay was developed by Choi and colleagues 43 to obtain differentiated myoblasts from iPSCs avoiding viral transfections and using xeno-free media, making iPSCs more suitable for clinical trials. 43
An in vitro human skeletal muscle model based on iPSCs could develop a biological platform to reach a deep understanding of muscle physiology, disease evolution, and drug efficiency or toxicity.
This platform could also represent a potential solution to the limits inherent in the use of animal models, which often present phenotypical and physiological differences in comparison with human disease.
However, some limitations remain, such as the fact that a single model could not portray all the phenotypes characterizing DMD and BMD patients, since they have a high clinical heterogeneity due to over 1000 different mutations in the dystrophin gene that give rise to great variability in patient clinical manifestations and responsiveness to therapeutic treatments. 58 Moreover, the application of iPSCs in skeletal muscle engineering introduces the intrinsic variability among patient-derived lines. At present, little has been done to characterize this heterogeneity, which is an obstacle in the employment of patient-based iPSC models, because it might hinder real comparisons among different cell lines, limiting the general findings on efficacy of possible therapeutic drugs. For this reason, the CRISPR/Cas9 technique has been introduced to genetically engineer aimed mutations in healthy donor iPSC lines. 139 Actually, the gene correction using the CRISPR-Cas9 system holds great promise for various applications, such as the study of gene functions, disease modeling and gene therapy, even if the safety in terms of mutagenesis of this approach remains unknown. While many different cell lines have been created using CRISPR-introduced mutations, only a few disease-causing mutations (e.g. null alleles) have been chosen and tested for therapeutic implications. Conversely, competent transfection methods and accurate detection assays to verify genomic cleavage are crucial for efficient genome editing in fragile iPSCs in order to obtain a suitable number of clones. 139 In addition, the creation of a set of cell lines characterized by different mutations in the dystrophin gene using the CRISPR/Cas9 technology would entail an increase in associated costs.
To reduce the risk for insertional mutagenesis and to correct large deletions, mini chromosomes, such as HACs are alternative strategies, because they remain permanently episomal in the host genome, replicate and segregate during mitosis and carry large genetic loads. 133
In the last few years, some skeletal muscle 3D models have been developed in order to study molecular phenotypes in neuromuscular disorders. One of the main aspects to consider when creating 3D models is that skeletal muscle myotubes are highly influenced by the surrounding tissue, and therefore, the substrate elasticity is important to mimic the native skeletal muscle environment, to enhance satellite cell proliferation and to differentiate myoblast precursors. 46
The most significant challenge is the development of fully matured skeletal muscle fibers to adequately model the adult physiological architecture. To date, different architectural scaffolds based on Matrigel, collagen, fibrin, or synthetic biopolymers have been tested successfully, providing the needed tension for final maturation, but it is still unclear how the different matrices contribute to reproducing the muscle architecture influencing myogenic activity in culture. 112 At present, some strategies are used to obtain complete maturation of engineered constructs that are equal to that of adult human muscle tissue and consequently, combinatorial methods seem to be necessary. Co-cultures with motor neurons and treatment with broad-field electrical stimulations are strategies that are able to induce the generation of directed forces in bioartificial muscles.103,104 In addition, these models can be further optimized by inserting other isogenic cellular types such as pericytes, endothelial cells and muscle interstitial cells, ameliorating the functionality and allowing the implantation of these engineered muscles. 107 Nevertheless, a univocal combination of electrical, chemical and biological stimuli that are necessary and sufficient to mimic native skeletal muscle complexity for disease modeling and drug screening must still be defined. Because different cellular types need specialized culture conditions and because some media used to culture nonmyogenic cells can interfere with transgene-free or small molecule treatments adopted to differentiate iPSCs, a combination of transgene-free and transgene-based methods seems to be required.35,42,107 Therefore, adaptation of these systems will be of fundamental importance in producing reliable skeletal muscle models for high volume drug screening testing. Recently, to implement the preclinical drug developments, Agrawal and co-workers created an effective tool, based on a chip system, that assumed the architectural and structural properties of skeletal muscle within a microfluidic device. 154
In recent years, efforts have also been made to achieve a model of cardiomyopathy beside skeletal muscle pathology, since cardiac dysfunctions frequently develop in patients affected with DMD and BMD disorders. Cardiomyocytes differ from skeletal muscle cells in terms of both dystrophin mRNA levels and turnover. Additionally, drug half-life in these cells is longer even if the delivery seems to be less efficient. 148 Moreover, primary human cardiomyocytes cannot be obtained easily or divide in culture, and it is therefore important to develop reliable in vitro models in order to obtain CMs from human iPSCs. Pluripotent stem cell-derived CMs are more similar to fetal CMs than adult CMs, and this immaturity promotes their use in drug screening and cell-based therapies. As extracellular matrix and 3D structure influence cellular behavior and maturation, assays have been developed to engineer cardiac tissue on scaffolds. 114 Furthermore, these new methods need endothelial cells, smooth muscle cells, myofibroblasts, mesenchymal stem cells and cardiac fibroblasts, which are important for contractile force improvements and producing angiogenic factors essential for a stable vessel maturation in vitro. 115 In addition, nanotechnologies have been created to check and record the cardiac microtissue-generated forces in real time, using mini-engineered heart tissue grown into microgauges. 117 However, engineered cardiac tissue is not fully developed for efficient drug screening analysis; the major hurdle to the application of these technologies is the inability to assess cell variability as well as chemical, mechanical, and electrical stimuli.
In conclusion, all these 3D skeletal and cardiac models in vitro show a correspondence with the structural and functional characteristics of muscle and heart tissue in vivo. These engineered constructs encompass key disease features of DMD and BMD, including inflammation and a scarce regenerative myogenic capacity, that are partially rescued by genetic and pharmacological therapies. Therefore, these approaches could provide useful platforms to implement future therapeutic strategies for these disorders and pave the way to potential high-throughput, low-volume drug screenings and the testing of efficacy and toxicity. Furthermore, since these models use iPSCs carrying patient-specific mutations, it will be possible to more accurately evaluate the effects of experimental drugs on different patients, eventually providing personally tailored therapies.
Footnotes
Acknowledgements
We gratefully acknowledge Centro Dino Ferrari for its support. Daniela Piga and Sabrina Salani contributed equally to this work.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.