
Abstract
The inflammatory response is a major factor in stroke pathophysiology and contributes to secondary neuronal damage in both acute and chronic stages of the ischemic injury. Recent work in experimental cerebral ischemia has demonstrated the involvement of neurotransmitter signaling in the modulation of neuroinflammation. The present review discusses recent findings on the therapeutic potential and diagnostic perspectives of cholinergic, purinergic and glutamatergic receptors and transporters in experimental stroke. It provides evidence of the role of neurotransmission signaling as a promising inflammatory biomarker in stroke. Finally, recent molecular imaging studies using positron emission tomography of cholinergic receptors and glutamatergic transporters are outlined along with their potential as novel anti-inflammatory therapy to reduce the outcome of cerebral ischemia.
Introduction
The ischemic injury is a complex system of pathological processes including excitotoxicity, oxidative stress, inhibition of protein synthesis, programed cell death and inflammation, among others, which occurs within minutes until hours and even days and months after the brain vessel occlusion. 1 This pathological process is initiated as a result of the impairment of energy levels that maintain ionic gradients, 2 inducing a loss of membrane potential in neurons and glia, a process known as anoxic depolarization (AD). 3 AD spreads as a self-propagating wave-like depolarization across the susceptible brain parenchyma due to the release of K+, glutamate and adenosine triphosphate (ATP). 4 As a result, increased entry of Ca2+ into the cell is thought to initiate a cascade of cytoplasmatic and nuclear events, including activation of proteolytic enzymes, production of reactive oxygen species (ROS), lipid peroxidation and membrane damage, the inhibition of protein synthesis, cerebral edema formation, cellular DNA fragmentation, and activation of apoptotic cell death which together lead to primary ischemic damage and the subsequent activation of inflammation. 5 The inflammatory response plays a pivotal role in exerting both beneficial and detrimental effects on the acute ischemic injury and the functional recovery after stroke. 6 In this review, we focus on the evidence regarding inflammatory biomarkers and cells that control the neuroinflammatory reaction and related mechanisms after stroke. In particular, the recent findings on the potential therapeutic and diagnostic perspectives of cholinergic, purinergic and glutamatergic biomarkers for the use in both therapy and diagnosis of stroke outcome will be also discussed.
Inflammation after brain ischemia
Inflammation is a complex response to necrotic cells and the subsequent generation of ROS during the secondary injury following stroke. 7 Early after ischemia, the release of a repertoire of different proinflammatory prostaglandines, cytokines and chemokines results in the activation of microglia, the resident immune cell population of the brain. 8 Once activated, microglia leads to the induction of adhesion molecules, integrins and selectines in both the brain vasculature and immune cells through the release of proinflammatory cytokines. 9 Hence, the overexpression of all these molecules mediates leukocyte adhesion to the vascular endothelium leading to subsequent entry into the brain tissue. 10 In addition, leukocyte infiltration is amplified by the disruption of the blood–brain barrier (BBB) through the release of cytotoxic agents such as metalloproteinases by microglia and other infiltrated leukocytes. 7 Microglia and infiltrated macrophages can be classified into at least two subsets with distinct molecular phenotypes and functions depending on the activation pathway. The ‘classically activated’ proinflammatory microglial cells and macrophages play a central role in host defense against pathogens, but they can also damage healthy cells as neurons and glial cells. In contrast, anti-inflammatory phenotypes or ‘alternatively activated’ cells downregulate inflammation and promote tissue remodeling or repair and angiogenesis after stroke. 11 Thus, a plethora of pathways and mediators might determine the final fate of these cells under a pathological situation in the brain. 12 Both ex vivo and in vivo studies have suggested that many neurotransmitter receptors including cholinergic, purinergic and glutamatergic receptors or transporters are overexpressed in microglia under pathological situations, behaving as modulators of the inflammatory response 12 (Table 1). Therefore, the precise management of neuroreceptors as inflammatory biomarkers may ultimately promote novel therapeutic and diagnostic strategies for treating ischemic damage.
Inflammatory biomarkers targeting cholinergic, purinergic and glutamatergic systems in stroke.
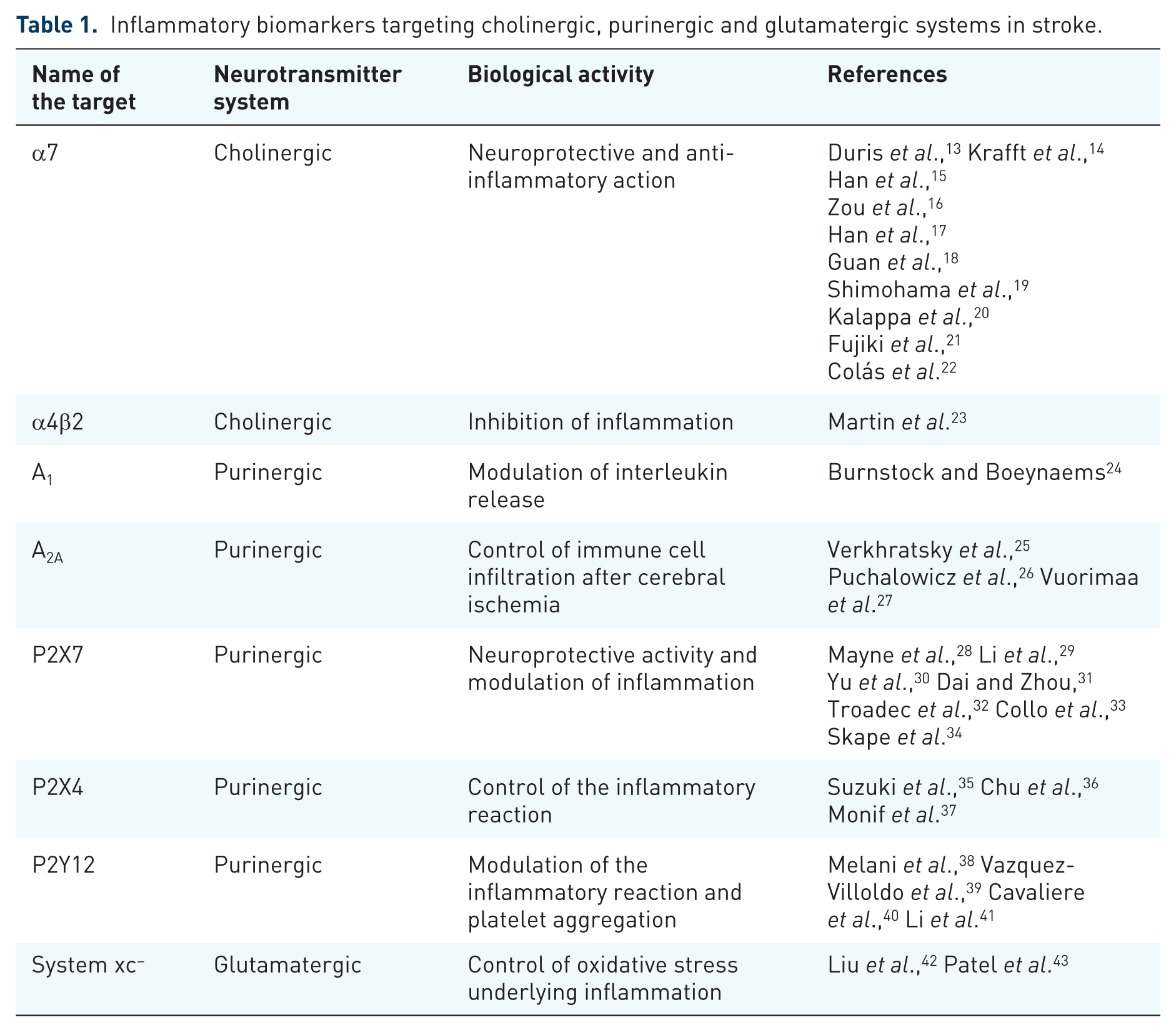
Cholinergic receptors
Cholinergic receptors show neuronal protective effects which have been related to the modulation of immune cells by the cholinergic anti-inflammatory pathway. 44 The mechanism for inhibition of cytokine release is attributable to acetylcholine (Ach) through the inflammatory reflex of the vagus nerve.45–47 Macrophages and other immune cells express acetylcholine receptors (AChRs), which are able to transduce an intracellular signal that inhibits cytokine synthesis. 48 To date, the most characterized cholinergic receptors as inflammatory modulators are those formed by the α7 subunit of the AChR. Studies in mice have shown that activation of these receptors is required for acetylcholine inhibition of macrophage tumor necrosis factor (TNF) release, becoming an essential key for the regulation of inflammation. 48 In addition, pharmacological stimulation of α4β2 nicotinic receptors promotes inhibition of amyloid toxicity to cortical neurons and modulation of inflammation after cerebral ischemia in rats.23,49
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels composed of five subunits (α2–α10 and β2–β4) and the most abundant nAChRs in the mammalian brain are heteromeric receptors containing α4 and β2 subunits and homomeric α7. 50 The expression of nAChRs has been described in a large variety of neural cells, 51 and non-neuronal cells such as microglia, astrocytes, oligodendrocyte precursor cells and endothelial cells,52–56 where they can decrease the extent of cell death and enhance synaptic plasticity. 57 Therefore, these receptors are potential therapeutic targets for several neurodegenerative disorders such as Parkinson’s disease, schizophrenia, depression and Alzheimer’s disease. 58 nAChRs have been proposed as promising novel candidates for the treatment of neuroinflammation following stroke. 57
As previously discussed, the cholinergic anti-inflammatory pathway is controlled by vagus nerve stimulation and particularly via the α7-nAChRs expressed on innate immune cells, evidencing the effect of the peripheral cholinergic system. 48 Some studies have shown that stimulation of the vagus nerve attenuates cerebral ischemia injury and reperfusion.59,60 A brief stimulation of the vagus nerve after both permanent and cerebral ischemia displayed a reduction in the protein levels of α7 receptors followed by a reduction on inflammation, apoptosis and neuroprotection through the α7-nAChR/JAK2 anti-inflammatory pathway.59,60 Accordingly, the pharmacological activation of α7 receptors with selective agonists confirmed reduction of the brain injury and neuroprotection after intracerebral hemorrhage in rodent models through reduction of the inflammatory response.13,14 In the latter study, the use of methyllycaconitine as a potent and selective α7-nAChR antagonist reversed the antinflammatory potential of the agonist PHA-543613 after intracerebral hemorrhage in mice. 14 Following cerebral ischemia, several preclinical studies have observed that both the local upregulation and the pharmacological activation of nicotinic receptors protects the brain against ischemic injury, suggesting the protective central cholinergic effect and the potential role of these receptors as promising inflammatory biomarkers.15–23
A novel approach proposed the use of positive allosteric modulators of α7 nAChRs that converts endogenous agonists of α7 nAChRs such as ACh into potent neuroprotective agents in postischemic neuronal injury in cortical and subcortical brain regions. 20 In another study, the use of donepezil, an acetylcholinesterase inhibitor used for the treatment of Alzheimer’s disease, displayed upregulation of nAChRs that attenuated the cerebral brain infarction volume after cerebral ischemia in rats and mice.21,61 Moreover, the use of other cholinesterase inhibitors such as huperzine A and galantamine has shown their potential anti-inflammatory and neuroprotective effects after cerebral ischemia in rodents.62–64 The pharmacological use of nicotine after a rat model of global ischemia increased the neuronal survival of CA1 pyramidal neurons accompanied by a reduction of microglial cells, TNFα and interleukin (IL)-1β in the region of the infarction. In addition, pretreatment with α-bungarotoxin, a selective α7 nAChR antagonist, could prevent the inhibitory effects of nicotine on cultured microglial proliferation, suggesting the role of nicotine in microglial activation through the activation of nicotinic receptors. 18 Therefore, these results suggest that cholinergic agonists may be of clinical relevance for the treatment of stroke.
The use of selective agonists of α7 nAChR such as PHA 568487 has also shown very promising results, reducing the ischemic brain injury and inflammatory response after experimental stroke in rodents.15,17,22 Following permanent cerebral ischemia in mice, treatment with PHA 568487 showed a reduction of functional deficits at the acute stage of cerebral ischemia. 15 Furthermore, PHA treatment reduced lesion volume, decreased the number of CD68+ and M1 macrophages, and increased the number of M2 or anti-inflammatory microglia or macrophages at days 3 and 14 after permanent middle cerebral artery occlusion (MCAO) in mice. 15 This study suggested that α7 receptors might decrease the inflammatory response through control over microglia or macrophage polarization after cerebral ischemia. Nevertheless, a recent study showed that the daily treatment of PHA 568487 during the first week after transient cerebral ischemia in rats displayed a nonsignificant decrease of the expression marker values for both proinflammatory and anti-inflammatory microglia markers. 22 Moreover, this study observed fewer values of selectines, adhesion molecules and infiltrated T lymphocytes after treatment with PHA, suggesting a possible role of α7 nAChRs in the regulation of leukocyte infiltration into the ischemic tissue. 22 Likewise, the infiltration of leukocytes after stroke can also be influenced by the disruption of the BBB; however, activation of α7 receptors showed similar levels of BBB disruption after MCAO in rats. 22 In contrast, Zou and colleagues examined the effect of α7 nAChRs activation with PHA after cerebral ischemia in mice showing an improvement of the BBB integrity. 16
In spite of all these discrepancies, α7 nAChRs play a promising key role in the inflammatory reaction following cerebral ischemia in rodents. For this reason, in vivo imaging of these receptors with a positron emission tomography (PET) technique might be of great importance to further our understanding of the role of α7 receptors in brain diseases such as stroke. During the last few years, promising radiotracers for imaging these receptors have been synthesized;65–69 however, only a PET imaging study has been carried out to evaluate the role of α7 nAChRs in neuroinflammation. This study used the selective orthosteric α7 nAChR agonist PET radioligand, [11C]NS14492 to monitor the expression of α7 receptors during the following month after cerebral ischemia in rats. PET imaging with [11C]NS14492 described an overexpression of these receptors as a response to the ischemic injury that was identified in microglia and infiltrated macrophages at day 7 after ischemia (Figures 1 and 2). Finally, PET imaging of neuroinflammation with [18F]DPA-714, a specific radioligand for the translocator protein (18KDa) (TSPO), 70 showed a reduction in the radioligand uptake as a result of treatment with PHA 568487 on day 7 after cerebral ischemia, supported by a reduction in activated microglia and macrophages 22 (Figure 2). Hence, the anti-inflammatory activity exerted by the cholinergic system following stroke has been attributed to α7 nAChRs, although α4β2 nicotinic receptors have also been involved in the inflammatory reaction underlying cerebral ischemia in rats. 23 The expression of α4β2 nicotinic receptors is increased after microglia or macrophage and astrocyte activation during cerebral ischemia evolution. 23 In vivo PET imaging with 2[18F]-fluoro-A85380, a selective radiotracer for α4β2 nAChRs, showed a radioligand uptake increase at day 7 after ischemia followed by a progressive decrease later on (Figure 1). The α4β2 expression pattern is in accordance with the uptake increase of [11C]PK11195, a PET radiotracer for imaging inflammation. 71 Furthermore, treatment with the α4β2 antagonist dihydro-β-erythroidine hydrobromide (DhβE) caused an increase in [11C]PK11195 binding after cerebral ischemia in rats, evidencing the potential role of α4β2 nAChRs in the regulation of the neuroinflammatory response after stroke 23 (Figure 1).

Temporal evolution of system xc−, α4β2 and α7 receptor expression after cerebral ischemia in rats. nAChR, nicotinic acetylcholine receptor.

Magnetic resonance imaging (MRI) and positron emission tomography (PET) with [11C]NS14492 and [18F]DPA-714, selective radiotracers for α7 nicotinic acetylcholine receptors (nAChRs) and inflammation respectively. (A) [11C]NS14492-PET and MRI coregistered images at control (day 0), day 3 and day 7 after middle cerebral artery occlusion (MCAO). (B) MRI-T2W and PET images of [18F]DPA-714 before (day 1) and at day 7 after MCAO in controls and in rats treated with the α7 antagonist PHA.
Purinergic receptors
In the nervous system, ATP and its derivatives act as extracellular signaling molecules through a large variety of receptors known as purinergic receptors. 12 The effects of purines and pyrimidines are mediated through an extended family of purinergic receptors, which are classified as metabotropic P1 adenosine receptors (A1, A2A, A2B and A3), metabotropic P2Y and ionotropic P2X purinoreceptors. 72 Several investigations have reported the role of purinergic signaling following neurodegenerative diseases, epilepsy, neuropsychiatric disorders and stroke. 73 Likewise, several studies have proposed ATP, adenosine and purinergic receptors as promising biomarkers for stroke therapy. 74 Cerebral ischemia leads to the high increase of concentrations of both ATP and adenosine that can stimulate the purinergic receptors. 75 Indeed, the effect of ATP and adenosine after cerebral ischemia may be due to the interaction of purinergic receptors expressed on neurons, glial cells and peripheral inflammatory cells such as lymphocytes and neutrophils.24–27,76
P1 receptors
Infusion of adenosine to the rat brain is protective against cerebral ischemia as it reduces the infarct volume and improves the neurological outcome 77 due to mainly the response of the adenosine A1 receptors. 74 Moreover, these receptors modulate the expression of IL-10 release by immune cells after neonatal hypoxic ischemic brain injury. 78 Despite these findings, the role of A1 receptors on inflammatory reaction after stroke has been scarcely explored to date. However, the A2A adenosine receptors are expressed in innate (microglia, macrophages, mast cells and neutrophils) but also in adaptive immunity (lymphocytes) cells, supporting its control of the neuroinflammatory response. 24 Following cerebral ischemia, activation of A2A receptors reduced processes related to the infiltration of peripheral inflammatory cells such as chemotaxis, rolling, adhesion and transmigration. 79 Accordingly, low doses of the selective A2A agonist CGS21680 decreased the number of infiltrated granulocytes, microgliosis, astrogliosis and improved myelin organization in the injured lesion after cerebral ischemia in rats. 80 In agreement with this effect, treatment with CGS21680 attenuated both the infiltration of neutrophils and TNFα production after intracerebral hemorrhage. 28 Activation of these receptors with BAY 60-6583 inhibits tissue plasminogen activator (tPA)-induced hemorrhagic transformation, thus reducing brain swelling and lesion volume after experimental stroke. 29 In addition, treatment with BAY 60-6583 induced a decrease in metalloproteinase-9 activation, which is mainly involved in the permeabilization of the BBB. 29 Therefore, restoration of the BBB by A2A receptors can likely reduce the infiltration of leukocytes into the infarcted tissue after stroke. Conversely, selective inactivation of A2A receptors in bone marrow-derived cells (BMDCs) protected against brain injury by reducing the production of proinflammatory cytokines such as IL-1β, IL-6 and IL-12 in the brain. 30 Hence, all these findings support the bidirectional modulation of inflammation by A2A receptors after cerebral ischemia. 31 Finally, activation of adenosine A3 receptors displayed an anti-inflammatory effect and a depletion of the infiltration or migration of macrophages and microglia after cerebral ischemia in rats. 81
P2 receptors
P2 receptors are classified in P2X ionotropic and P2Y metabotropic receptors. P2X receptors are a family of seven receptors (P2X1-7) permeable to cations (Na+, K+ and Ca2+) and expressed on the surface of cells. P2Y receptors are coupled to G protein and a total of nine subtypes P2Y1,2,4,6,11,12,13,14 have been cloned to date in mammalian species. 72 An increase in the extracellular concentrations of ATP after brain injury results in direct damage to neurons and oligodendrocytes through Ca2+ intracellular loading, a result of P2X7 activation.82,83 Thus, P2X7 antagonists reduce neuronal damage and infarct size after transient focal ischemia32,83,84 and ameliorate oligodendroglial and axonal damage after white matter ischemia. 82 Furthermore, ATP might also be crucial for the regulation of microglia after pathological situations. 33 In the early stages of ischemia, low ATP levels can induce the recognition and migration of microglia to the lesion. 34 In fact, activation of microglia from their resting state is marked by overexpression of P2X7 receptors in the peri-infarct region, which promotes the release of neurotrophic factors and the enhancement of neuronal survival.35,85 However, during the later stages of cerebral ischemia the ATP levels increase, promoting the proliferation of microglia and cell death. 36 Under these conditions, microglia upregulate the expression of proinflammatory cytokines such as IL-1β, IL-6 and TNFα, enhancing the inflammatory response after cerebral ischemia. 37 In addition, de novo expression of P2X7 receptors observed in both activated and reactive microglia suggests a differential role of these receptors in core and neighboring regions of the brain infarction. 38 The P2 unselective antagonist Reactive Blue reduced ischemic brain damage by blocking activated microglia in the core of the lesion. Conversely, the same treatment increased the expression of P2X7 receptors in remote areas, promoting the restoration and defense of the tissue. 38 Therefore, these temporal and spatial contrary processes should be taken into account in future therapeutic approaches targeting these receptors for treating stroke.
Microglial cells have also been characterized by the overexpression of P2X4 receptors that contribute to the control of the fate and the survival of the microglia. 39 Following brain ischemia, P2X4 receptors are upregulated in microglia or infiltrated macrophages and P2 antagonists can decrease ischemic cell death, reducing P2X4 receptor expression. 40 During hypoxic situations, expression of these receptors mediates activation of the amoeboid microglial cells and the release of proinflammatory cytokines induced by the increase in ATP levels. 41 Therefore, P2X4 receptor expression on microglial cells is associated with the progression of neuroinflammation following cerebral ischemia, despite the underlying mechanisms not being clearly deciphered to date. 86
P2Y12 receptors have also been shown to be expressed on microglial cells, suggesting their role in inflammation.87,88 The oral administration of the P2Y12 antagonist ticagrelor promotes protection against stroke damage through the inhibition of microglia activation, infiltration of blood-derived cells and the expression of proinflammatory mediators such as IL-1, monocyte chemoattractant protein 1 (MCP-1) and nitric oxide synthase (iNOS). This study also showed that ticagrelor inhibited adenosine diphosphate (ADP)-induced chemotaxis in primary cultured microglia. 88 Furthermore, ticagrelor exerts antithrombotic actions and the P2Y12 receptor is also expressed on circulating platelets, mediating its aggregation and activation. 89 Moreover, the use of clopidogrel, a P2Y12 receptor inhibitor with antiplatelet action, has been used in therapy for the secondary prevention of ischemic stroke. 42 These findings suggest that P2Y12 antagonist can conduct anti-inflammatory, neuroprotective and antiplatelet activity after cerebral ischemia. 74
Glutamate transporters
Although glutamate is the main excitatory neurotransmitter in the central nervous system (CNS), activation of glutamate receptors induces neuronal death after stroke. 90 Under physiological situations glutamate is stored intracellularly, however under pathological conditions such as cerebral ischemia the levels of extracellular glutamate can dramatically be increased, promoting the excitotoxicity mechanism through the influx of calcium into the neurons. 91 The clearance of extracellular glutamate levels through a family of transporter proteins called excitatory amino acid transporters (EAATs) has been mainly attributed to the glutamate-scavenging action of astrocytes. 92 In the normal CNS, astrocytes express EAAT1, EAAT2 and glutamine synthetase (GS) that first transport glutamate into intracellular milieu, and second convert the glutamate into glutamine. 93
Cystine glutamate antiporter
Another transporter involved in glutamate homeostasis is the cystine glutamate antiporter, also known as system xc−. 94 It is a heterodimer composed of a heavy chain subunit (4F2hc) and a light chain subunit (xCT). 95 System xc− mediates the cellular import of cysteine and the release of glutamate to the extracellular space in exchange. Cystine is intracellularly converted to cysteine, the rate-limiting substrate for gluthatione aproduction and oxidative protection. Thus, the expression and function of system xc− are modulated in different pathologies secondary to the induction of oxidative stress.96–98
After cerebral ischemia, overexpression of the cystine glutamate antiporter contributes to the increase in the extracellular glutamate concentration that promotes ischemic neuronal death through activation of extrasynaptic N-methyl-D-aspartate (NMDA) receptors 95 (Figure 3). This study shows that pharmacological inhibition of system xc− displays reduced neuronal death after in vitro ischemia. 95 Furthermore, in vivo PET imaging of system xc− activity with the radiotracer [18F]FSPG showed an increase in the PET signal uptake from 5 min to 5 h after ischemia, showing the increase in the function of these transporters during the subacute stage of stroke. 95 [18F]FSPG is a fluorine-18-labeled L-glutamate derivative taken by the system xc− due to the lack of discrimination between its natural substrate cystine and glutamate for the inward transport.43,99 Recently, a PET study with this radiotracer has demonstrated the overexpression of system xc− on microglial cells following experimental autoimmune encephalomyelitis (EAE) in rats. 100 Additionally, the depletion of microglia with clodronate showed a reduction in the [18F]FSPG PET signal in the spinal cord, confirming the link between microglial activation and cysteine or glutamate antiporter activity in EAE rats. 100 Another clinical study used PET with [18F]FSPG to detect inflammatory lesions provoked by the activation of macrophages in diseases such as sarcoidosis. 101 Several studies support these findings showing that the expression of system xc− is particularly abundant in microglia, macrophages and other immune cells.102,103 In fact, the release of glutamate from both resting and activated microglia by system xc− can induce a regional increase in glutamate levels that leads to excitotoxic oligodendrocyte death contributing to the pathogenesis of white matter disorders. 104

The release of glutamate during brain ischemia triggers neuronal death by overactivation of NMDA receptors. Different mechanisms contribute to glutamate homeostasis alterations. Astrocytic glutamate transporters play a key role in maintaining synaptic glutamate levels. However, extrasynaptic glutamate is mainly regulated by the cystine glutamate antiporter, also known as system xc−. 94 Because N-methyl-D-aspartate (NMDA) receptors involved in neuronal death are typically extrasynaptic, it has been proposed that glutamate release by the cysteine or glutamate antiporter activates extrasynaptic N-methyl-D-aspartate receptors (NMDARs). 95 Cells involved in glutamate release by cystine glutamate antiporter during ischemic insults remain to be determined. EAAT, excitatory amino acid transporter.
Upregulation of system xc− occurs during the following week after ischemia reperfusion 104 (Figures 1 and 4). This stands in accordance with the expression of TSPO, a marker or microglial activation, using the radiotracer [18F]DPA-714. 105 Indeed, the expression of the cysteine/glutamate antiporter takes place in activated microglia and infiltrated macrophages during the first week, and marginally in astrocytes at 1 month after ischemia onset. Additionally, the inhibition of system xc− with sulfasalazine and S-4-CPG resulted in a decrease in inflammatory response through the inactivation of both microglia and infiltrated macrophages, a decrease in proinflammatory markers (CCL2, TNF and iNOS) and an increase in the anti-inflammatory marker arginase after experimental stroke in rats. 105 These results showed that the blocking of the source of glutamate release on microglia and macrophages during cerebral ischemia evolution may be a relevant therapeutic intervention to halt progression of the brain damage after ischemia. Furthermore, these results fully support that system xc− might play a key role in the modulation of inflammatory response following stroke.

Magnetic resonance imaging (MRI) (T2 weighting (T2W)) and positron emission tomography (PET) with [18F]FSPG and [18F]DPA-714, markers of cystine glutamate antiporter activity and inflammation respectively. (A) [18F]FSPG-PET and MRI coregistered images at control (day 0), day 3 and day 7 after middle cerebral artery occlusion (MCAO). (B) PET images of [18F]DPA-714 at day 7 after cerebral ischemia in vehicle (MCAO), SAS (MCAO+SAS) and S-4-CPG (MCAO+ S-4-CPG) treated rats. SAS and S-4-CPG are inhibitors of cystine glutamate antiporter.
Conclusion
Inflammation plays an important role at different stages of the cerebral postischemic injury. Consequently, the use of anti-inflammatory strategies in stroke therapy might offer a wider therapeutic window than current treatments. The inflammatory reaction following stroke involves a large variety of signaling pathways and mediators that can determine stroke outcome. During the last decade, both in vivo and ex vivo studies have described the role of neurotransmitter receptors and transporters, including cholinergic, purinergic and glutamatergic on the modulation of the inflammatory reaction after cerebral ischemia. It became evident that the expression of these receptors and transporters in neurons, glial cells and infiltrated immune cells in the ischemic brain promotes the release of a battery of signal molecules that enhance the inflammatory response after cerebral ischemia. Moreover, the activation or inhibition of these neuroreceptors and transporters has shown promising therapeutic responses in animal models of stroke. Recently, molecular imaging studies using PET of these neuroreceptors have described their link with the inflammatory reaction after ischemia and their potential role as novel imaging biomarkers of neuroinflammation. Therefore, the data reviewed here suggest that cholinergic, purinergic and glutamatergic agents may be useful both as biomarkers for neuroinflammation and as a treatment to attenuate the deleterious consequences of the inflammatory response after stroke. In our opinion, among the different candidates proposed here, the activation of nicotinic receptors might become a promising strategy for treating stroke in the near future. Despite this, future clinical studies are needed to support all these findings and their true potential to ameliorate the neurological care of stroke.
Footnotes
Funding
We acknowledge financial support by MINECO SAF2014-54070-JIN (A.M.) and SAF2016-75292-R (C.M.).
Conflict of interest statement
The authors declare that there is no conflict of interest.