
Abstract
Acute stroke is one of the major causes of death and disabilities. Since the 1980s many clinical studies have been conducted to evaluate neuroprotective approaches to treat this important brain vascular event. However, to date the only drug approved (recombinant tissue plasminogen activator [rtPA]) represents a thrombolytic, nonneuroprotective approach. An important neuroprotective strategy is based on erythropoietin (EPO). Exogenously administered EPO exhibits neuroprotective effects in numerous animal models, through the activation of anti-apoptotic, anti-oxidant and anti-inflammatory pathways as well as through the stimulation of angiogenic and neurogenic events. The capability of EPO to cross the blood–brain barrier after systemic administration and its effective therapeutic window are advantages for human acute stroke therapy. However, a multicenter stroke trial where recombinant human EPO (rhEPO) was combined with rtPA had negative outcomes. The present paper reviews the EPO neuroprotective strategy and its mechanisms in ischemic stroke and in other human nervous system diseases.
Introduction
Stroke is the third major cause of death in the most industrialized countries after cardiovascular disease and cancer. The overall incidence of stroke is predicted to increase over the next decade by 12%, although for low-income families this figure will be around 20%. More than 30% of the stroke survivors will have severe disabilities [Green, 2008; Shuaib and Hussain, 2008].
Strokes in the Western world are mainly ischemic (85%), resulting from an occlusion of a major cerebral artery, commonly the middle cerebral artery, by a thrombus or embolism [Green, 2008]. The rest of the strokes are hemorrhagic, where a blood vessel bursts either inside the brain or on its surface.
An effective pharmacological treatment immediately after an ischemic stroke to lessen cerebral damage remains an elusive goal. The recombinant tissue plasminogen activator (rtPA) is the only drug currently approved for clinical use in most countries. However, its short therapeutic window (4.5 hours), the risk of cerebral hemorrhage and the need for imaging studies to confirm the ischemic nature of the event, are among the reasons why this thrombolytic agent is only given to 4–5% of the patients [Green, 2008].
The first clinical studies to evaluate neuroprotection strategies began in the 1980s. To date, more than 1000 compounds, effective in animal models of acute ischemic stroke have been developed. Unfortunately, almost none of them has been approved for clinical use [O’Collins et al. 2006]. Although these failures have over shadowed the neuroprotection approaches, there are some promising neuroprotective agents such as citicoline and erythropoietin (EPO). Citicoline is an intermediary in the phosphotidylcholine biosynthesis with a role in regulating cell membrane integrity [Conant and Schauss, 2004]. A recent drug surveillance study in acute ischemic stroke patients showed that oral citicoline improved neurological, functional and global outcomes [Cho and Kim, 2009].
EPO, originally described as an erythroid differentiator cytokine [Fisher, 2003], has also been extensively studied as a tissue protective agent. In addition to its known role in erythrocyte formation, its functions in the central nervous system came out after the finding of EPO and EPO receptor (EPO-R) expression in neuro-epithelial tissues [Marti et al. 1996]. Hypoxia and other forms of brain injury upregulate the expression of EPO and its receptor in the nervous tissue [Marti, 2004]. There are several in vitro and in vivo studies reporting neuroprotective effects of EPO [Brines and Cerami, 2005; Genc et al. 2004; Marti, 2004].
The first clinical study in acute ischemic stroke using recombinant human EPO (rhEPO) was published in 2002. High doses of rhEPO proved to be safe and effective after 3 days of treatment [Ehrenreich et al. 2002]. The next report, which appeared 7 years later, had negative results: there was a higher death rate in patients receiving rhEPO and there were no benefits in imaging or clinical outcomes [Ehrenreich et al. 2009].
The aim of this article is to review the EPO neuroprotective strategy in ischemic stroke and in other human nervous system diseases.
Erythropoietin
EPO is a 165 amino acid glycoprotein produced mainly by peritubular cells in the adult kidneys and by hepatocytes in the fetus [Fisher, 2003]. EPO acts on the later stages of erythroid progenitor cells development, allowing maturation of erythroid precursors by inhibiting programmed cell death (apoptosis) and thus regulating red cell production [Fisher, 2003]. rhEPO is currently effective and widely used to treat different types of anemia, not only in uremic patients but also in newborns with anemia of prematurity, in patients with cancer-related anemia or myeloproliferative disease, thalassemias, bone marrow transplants, or those with chronic infectious diseases [Buemi et al. 2003].
The finding of the functional expression of EPO and its receptor in the nervous system of rodents, primates and humans, their expressions in neurons, glial and endothelial cells, together with the mounting evidence of EPO’s neuroprotective effects, expanded the idea of the biological function of EPO beyond erythropoiesis [Bernaudin et al. 2000; Marti et al. 1996; Sirén et al. 2001].
Marked changes in EPO and EPO-R gene expressions have been reported to occur in neurons, endothelial cell and glial cells after ischemic injury [Bernaudin et al. 1999, 2000; Marti et al. 1996; Sadamoto et al. 1998]. The weak constitutive expression of EPO in the adult brain can be rapidly increased by hypoxia and acute metabolic stress as evidenced by its detection in human cerebrospinal fluid or in postmortem brain tissue after traumatic brain injury, subarachnoid hemorrhage or stroke [Siren et al. 2009; Sirén et al. 2001; Springborg et al. 2003]. In addition, EPO-R is also upregulated in chronic brain diseases (e.g. schizophrenia and Alzheimer’s disease) possibly reflecting an ongoing metabolic distress [Ehrenreich et al. 2004; Siren et al. 2009].
In vitro EPO neuroprotective effects
The neuroprotective effects of EPO were first demonstrated in in vitro experiments, where mainly neuronal cultures have been challenged with several hypoxic or toxic injuries.
EPO protected hippocampal and cortical neurons from glutamate neurotoxicity in a dose-dependent manner, when applied 8 h prior to the insult; the protection was completely reversed by a co-application of a soluble EPO-R. These authors also showed that a short exposure to EPO (5 minutes) is enough to elicit the protective effect [Morishita et al. 1997]. EPO can also reverse hypoxia-induced hippocampal neuronal death when applied simultaneously with the hypoxic stimulus [Lewczuk et al. 2000].
In a hypoxia and glucose deprivation model, EPO protected cultured neurons, but not astrocytes from death, when applied at same time to the insults. Moreover, EPO reduced the excitotoxic effect of glutamate and a glutamate receptor agonist (AMPA: alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid) upon cortical neuron cultures [Sinor and Greenberg, 2000]. Pretreatment with EPO also prevented apoptosis induced by NMDA (N-methyl-d-aspartate) or by nitric oxide in neurons of cerebrocortical cultures [Digicaylioglu and Lipton, 2001].
It is reported that EPO increased PC12 cells survival when submitted to serum and nerve growth factor depletion [Koshimura et al. 1999] or to oxidative stress and apoptosis induced by 1-methyl-4-phenylpyridinium (MPP(+)) [Wu et al. 2007]. In a tissue culture model (hippocampal slices), EPO protected neurons from oxygen and glucose deficiency [Ruscher et al. 2002].
The effect of EPO on glial cell types has also been studied in vitro. The EPO/EPO-R system seems to participate in neural, but also in glial, cell development. In late-stage but still immature oligodendrocytes, EPO facilitates maturation as it increases the myelin basic protein expression and promotes oligodendrocyte processes formation. It also enhances astrocyte proliferation [Sugawa et al. 2002]. Pretreatment with EPO significantly attenuated multiple hallmarks of apoptotic cell death in astrocytes exposed to nitric oxide and staurosporine [Diaz et al. 2005]. The EPO-stimulated astrocytes may further accelerate the oligodendrocytes’ maturation. Finally, EPO maintained microglial integrity during oxidative stress induced by oxygen and glucose deprivation; less membrane phosphatidylserine exposure and less genomic DNA degradation were also evidenced [Li et al. 2006].
In vivo rhEPO neuroprotective effects
rhEPO has proven neuroprotective effects in several models of nervous tissue damage [Buemi et al. 2003; Genc et al. 2002]. Its ability to protect hippocampal neurons from delayed cell death, to reduce degenerating synapses in the ischemic hippocampal CA1 region and to prevent cognition impairment were the first neuroprotective effects reported for rhEPO in an in vivo global cerebral ischemia model in gerbils [Sakanaka et al. 1998].
Endogenous brain-derived EPO has been shown to be crucial for neuronal survival in vivo. Infusion of a soluble EPO-R into the brain of gerbils submitted to a mild ischemia that did not produce neuronal damage by itself, resulted in hippocampal neuronal cell death [Sakanaka et al. 1998].
The neuroprotective properties of rhEPO have also been demonstrated in focal permanent ischemia models, where it reduces both cortical infarct size and neurologic dysfunction [Bernaudin et al. 1999; Sadamoto et al. 1998]. A 6-hour therapeutic window and rhEPO’s capability to cross the blood brain barrier after systemic administration are among the advantages that could benefit human therapy [Brines et al. 2000; Wang et al. 2007].
The anti-apoptotic effect of EPO is one of its neuroprotective mechanisms. This effect has been evidenced by a decreased number of apoptotic cells [Chong et al. 2002; Digicaylioglu and Lipton, 2001; Wang et al. 2007] and by modulation of other molecules involved in the apoptotic pathways [Chong et al. 2002; Kilic et al. 2005; Renzi et al. 2002].
Other protective effects such as angiogenesis and neurogenesis could be exerted by rhEPO [Shingo et al. 2001; Wang et al. 2004a] (Figure 1). Starting treatment 24 h after stroke, rhEPO induced angiogenesis via upregulation of vascular endothelial growth factor (VEGF). rhEPO also induced capillary-like formations; the latter effect was blocked by a specific VEGF-receptor antagonist [Wang et al. 2004a].

Protective pleiotropic effects elicited after EPO binds EPO-R.
EPO is required for normal brain development in mammals [Juul, 2002; Yu et al. 2002]. It has been shown that EPO regulates neurogenesis in the adult mouse brain [Shingo et al. 2001]. rhEPO-induced neurogenesis has been studied in in vivo and in vitro experiments. Seven days after an embolic stroke, rhEPO increased the number of migrating neuroblasts in the ipsilateral subventricular zone and in the ischemic boundaries of the adult rat brain [Wang et al. 2004a]. These EPO effects possibly promote neural plasticity during stroke recovery, which emphasizes the regenerative potential of this molecule.
The preservation of the white matter is also critical for limiting and recovering functional deficits of motor and sensory functions. Following hypoxic stimulation in cultured astrocytes, there is an increase of endogenous EPO mRNA and EPO levels. Furthermore, endogenous astrocyte EPO/EPO-R could prevent oligodendrocyte progenitor cells damage under hypoxic/reoxygenation condition [Kato et al. 2011].
In addition, EPO amplifies stroke-induced oligodendrogenesis and axonal remodeling. Administration of rhEPO 24 h after embolic focal cerebral ischemia induced sustained oligodendrocyte progenitor cells proliferation in the peri-infarct white matter and in the subventricular zone [Zhang et al. 2010]. Six weeks after stroke, rhEPO-treated animals showed more mature oligodendrocytes and more myelinated axons in the peri-infarct white matter; these effects were associated with considerable functional improvement [Zhang et al. 2010]. Therefore, beneficial effects of EPO on glial cells may contribute to tissue regeneration and functional recovery after stroke.
EPO is also one of the mediators of ischemic and hypoxic preconditioning. In this protective phenomenon, an exposure to a brief, nontoxic episode of ischemia activates certain cellular pathways that finally increase the resistance of neurons to subsequent more severe insults [Genc et al. 2002]. Induction of EPO and VEGF expressions by hypoxia-inducible factor (HIF) is crucial for preconditioning [Bernaudin et al. 2002; Prass et al. 2003; Ruscher et al. 2002]. The EPO-R expression is necessary for the onset of an immediate preconditioning phase, and the reaction is limited to EPO-R positive cells at the time of exposure. Levels of both EPO and EPO-R increase significantly in a delayed preconditioning phase, with EPO-R expression upregulated first [Brines and Cerami, 2005]. The endogenous EPO/EPO-R system activated by neuroprotective mechanisms in neuronal injury could be reinforced by the administration of exogenous EPO [Genc et al. 2002].
EPO derivative molecules
Repeated rhEPO administrations could lead to potentially harmful increases in the red cell mass and platelet adhesion. Transgenic mice overexpressing systemic EPO developed larger infarcts than wild-type controls, indicating that rhEPO systemic chronic treatment might increase hematocrit and thus deteriorate the after-stroke outcome [Wiessner et al. 2001].
In order to circumvent that situation, EPO variants retaining the beneficial tissue-protective, but not the bone marrow-stimulating actions have been developed. Among the structurally related EPO derivatives, asialoerythropoietin (asialo-EPO), neuro-EPO and darbepoetin can be mentioned. Based on the fact that only a brief exposure is needed to achieve neuroprotection [Morishita et al. 1997], while the stimulation of erythropoiesis requires a sustained presence of EPO, these analogs differ from rhEPO in the oligosaccharide composition, as an attempt to modify its plasma half-life. Asialo-EPO is generated by total enzymatic desilylation of rhEPO with a very short plasma half-life in vivo. This very short-acting analog could have neuroprotective advantages in future clinical trials [Erbayraktar et al. 2003; Wang et al. 2004b].
Neuro-EPO is a variant with a low-sialic acid content and a short half-life. Intranasally administered Neuro-EPO exhibits neuroprotective effects in gerbil models of brain ischemia [Sosa et al. 2008; Subiros Martínez et al. 2007]. The use of the nasal route as a new delivery pathway to the brain is aimed to achieve neuroprotective concentrations in the nervous tissue as quick as possible using small drug doses. Drug transport from the nasal cavity directly to the brain has been shown to be feasible, even for challenging drugs such as small polar molecules, peptides and proteins, in animals and humans [Digicaylioglu, 2010; Graff and Pollack, 2005; Illum, 2003; Yu et al. 2005].
Darbepoetin (also known as ‘novel erythropoiesis stimulating protein’ [NESP]) is another EPO derivative [Siren et al. 2009]. This molecule contains additional oligosaccharide chains, thereby its circulation time is greater than rhEPO.
Another EPO derivative is the carbamylated EPO (CEPO). The carbamylation process transforms all lysines into homocitrullines which considerably modifies the protein conformation [Leist et al. 2004]. It has been suggested that CEPO binds to an heteromeric receptor complex, different from EPO-R in myeloid cells [Brines and Cerami, 2005]. The CEPO molecule, which is nonerythropoietic but fully tissue protective has shown similar efficacy as rhEPO for tissue protection in in vitro studies as well as in rat models of stroke, spinal cord compression, diabetic neuropathy and experimental autoimmune encephalomyelitis [Leist et al. 2004; Mahmood et al. 2007; Schmidt et al. 2008]. All of these derivatives possess at least comparable efficacy and potency and could offer important therapeutic advantages, without erythropoiesis stimulation.
A group of structural variants called EPO mimetic peptides (helix B peptide, Epopeptide AB and hematide) also has nonerythropoietic tissue-protective effects [Brines et al. 2008]. The induction of EPO gene expression has been the strategy to the development of other EPO variants (i.e. functional EPO variants). This is the case of HIF stabilizers and GATA-2 inhibitors, which are actively pursued as alternatives to EPO for erythropoiesis stimulation and ultimately also for neuroprotection [Siren et al. 2009].
EPO molecular mechanisms in brain protection
The molecular mechanisms by which EPO mediates its effects in the central nervous system are not completely elucidated. As EPO promotes cell viability by repressing apoptotic signals in bone marrow erythroid-precursor cells [Koury and Bondurant, 1990], several authors have proposed that similar mechanisms might operate in neuronal and endothelial cells [Buemi et al. 2003; Eid et al. 2004; Marti, 2004; Wang et al. 2004a].
The hematopoietic effect of EPO involves two EPO-R units homodimerization [Fisher, 2003; Siren and Ehrenreich, 2001]. It has been proposed that the neuroprotective effects of EPO are mediated by a heteromeric receptor complex composed of one EPO-R subunit and a dimer of the common beta-chain shared by the members of the interleukin-3 receptor family [Brines et al. 2004; Leist et al. 2004]. However, to date there is no solid proof for the existence of this heteromeric complex in neurons.
The binding of EPO-R with its constitutive ligand induces a conformational change that activates the EPO-R associated Janus tyrosine kinase 2 (JAK2) [Sola et al. 2005]. JAK-2 phosphorylation and activation leads to phosphorylation of several downstream signaling pathways that in neurons include: phosphatidylinositol-3-kinase (PI3K)/Akt, Ras/extracellular signal regulated kinase (ERK1/2), signal transducers and activators of transcription (STAT) [Siren and Ehrenreich, 2001], nuclear factor kappa-B (NF-κB), and calcium influx [Digicaylioglu and Lipton, 2001; Siren et al. 2009]. Finally, the expression of neuroprotective genes and apoptosis prevention are promoted [Juul, 2002; Siren et al. 2001] (Figure 2).
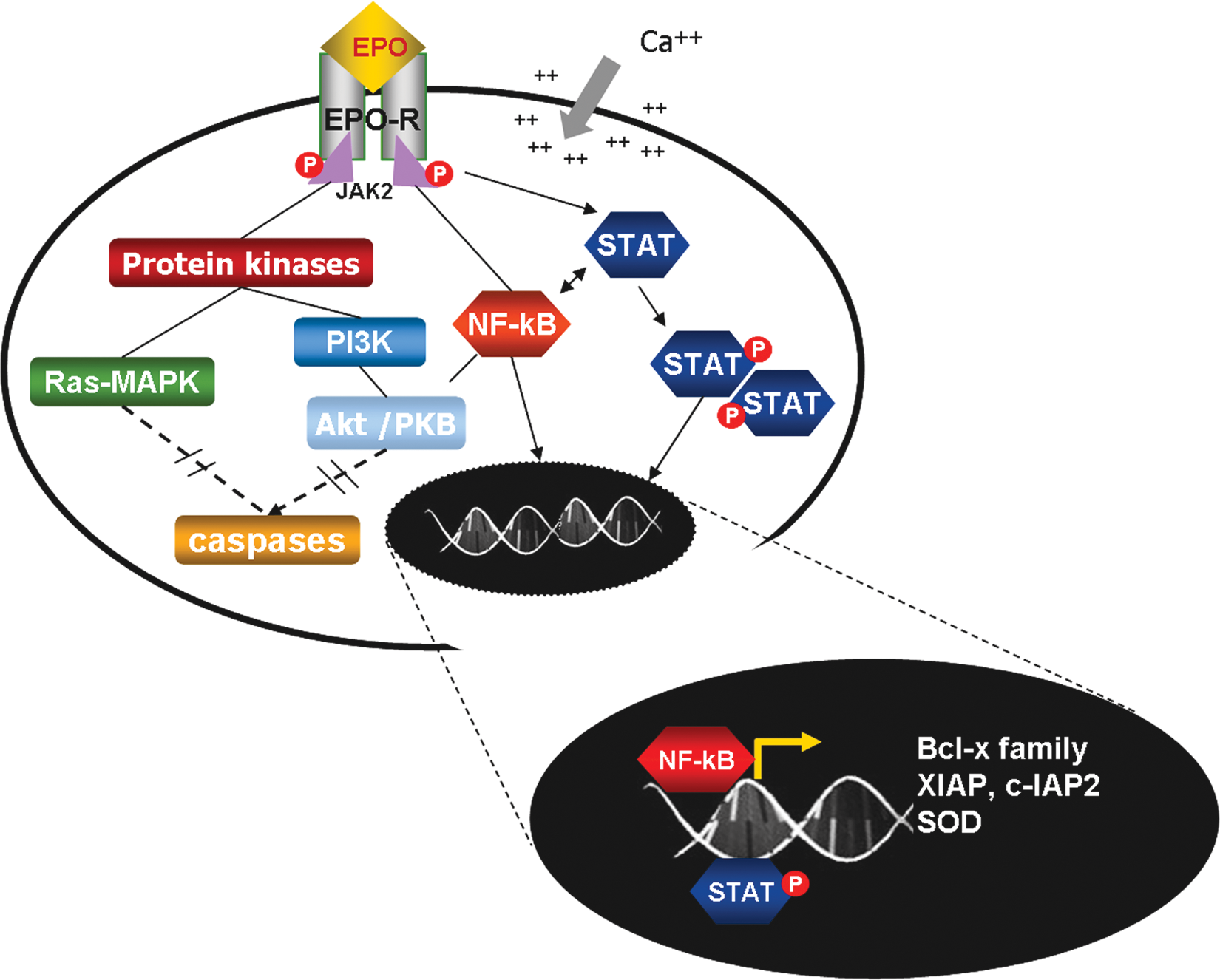
Intracellular signaling pathways triggered with the activation of EPO-R.The activation of the EPO-R induces several downstream signaling cascades which result in inhibition of caspase activation and antiapoptotic genes induction. JAK2: Janus tyrosine kinase 2; STAT: signal transducers and activators of transcription; PI3K-Akt/PKB: phosphatidylinositol-3-kinase-Akt/protein-kinase B; Ras-MAPK: Ras-mitogen-activated kinase; NF-κB: nuclear factor κB; xIAP and cIAP: x-linked and cellular inhibitors of apoptosis respectively; SOD: superoxide dismutase.
EPO may also repress cell death in neurons by activation of anti-apoptotic genes. Indeed, increased expression of apoptosis-inhibitor genes XIAP and c-IAP2 was demonstrated in cortical cells pre-incubated with rhEPO [Digicaylioglu and Lipton, 2001]. Furthermore, increased bcl-xL expression in the hippocampal CA1 region of ischemic gerbils was shown after rhEPO administration [Wen et al. 2002]. The anti-apoptotic effect of EPO has also been evidenced in an endothelial cell culture after an anoxic stimulus, where it could prevent DNA destruction, phosphatidylserine exposure and inhibit caspase 8-, caspase 1-, and caspase 3-like activities [Chong et al. 2002].
EPO has been shown to have beneficial effects against excitotoxic injury [Brines et al. 2000; Morishita et al. 1997], oxidative stress [Kumral et al. 2005] and inflammation [Agnello et al. 2002; Brines et al. 2000; Campana et al. 2006]. In the brain, EPO increases in response to oxidative or nitrosative stress [Bernaudin et al. 2000]. Protection against oxidative stress by EPO involves preventing excitotoxicity [Brines et al. 2000; Morishita et al. 1997] and free radical exposure [Calapai et al. 2000; Siren et al. 2001]. Activation of neuronal EPO-R prevents apoptosis induced by NMDA or by nitric oxide via triggering crosstalk between the signaling pathways of JAK2 and NF-kB [Digicaylioglu and Lipton, 2001].
The reduction of ischemia-induced inflammation associated to EPO is probably mediated by less neuronal cell death [Chen et al. 2007; Savino et al. 2006; van der Kooij et al. 2008]. This effect results in a decreased cerebral attraction of the inflammatory cells that would have produced the cytokines (IL-1beta, TNF-alpha and others) in unprotected animals [Villa et al. 2003].
Iron has also been considered an inflammation-supporting agent. Increased body iron stores have been related to greater oxidative stress and brain injury in clinical and experimental cerebral ischemia and reperfusion [Millan et al. 2008]. EPO treatment leads to temporary shifts in iron stores, leading to laboratory results similar to real iron deficiency [Siren et al. 2009]. However, iron substitution would induce a thrust of red blood cell production, definitely undesirable in nervous system indications. So, during EPO treatment no iron substitution should be allowed and the hematocrit has to stay within clearly defined limits. With cessation of the EPO treatment, most iron parameters will rapidly return to normal levels [Ehrenreich et al. 2007].
In cerebral white matter injury, the expression and activation of Akt1 is vital for EPO’s cytoprotective capacity, since pharmacological inhibition of the PI 3-K pathway or gene silencing of Akt1 expression eliminates the ability of EPO to protect microglial cells in vitro [Li et al. 2006]. In addition, in two models of periventricular leukomalacia (a neuropathological condition characterized by cerebral white matter impairment), EPO treatments decreased microglia activation, oligodendrocyte damage and myelin depletion. These effects were associated with decreased poly-(ADP-ribose) polymerase-1 (PARP-1) activity [Liu et al. 2011]. Furthermore, protection provided by EPO against oxidative stress induced by oxygen and glucose deprivation to a microglial cell line was closely associated with the inhibition of glycogen synthase kinase-3beta activity [Li et al. 2006].
Stimulation of neurogenesis and astrocytic differentiation from neuronal stem cells by EPO in vitro was dependent on nuclear translocation of NF-κB [Shingo et al. 2001].
Adhesion of preclinical EPO studies to STAIR criteria
The efficacy of rhEPO and its analogues regarding infarct size reduction (32%) and poststroke functional recovery in experimental focal cerebral ischemia is confirmed by a meta-analysis: higher doses of rhEPO were associated with smaller infarct volumes and were more effective when treatment started within the first 6 hours after stroke onset. However, when considering common bias sources as randomization and blindness, effectiveness falls [Minnerup et al. 2009]. The latter authors also evaluated whether the preclinical EPO studies included in the meta-analysis met the updated STAIR (Stroke Therapy Academic Industry Roundtable) recommendations (Table 1) [Fisher et al. 2009; Fisher, 2011].
Updated Stroke Therapy Academic Industry Roundtable (STAIR) preclinical recommendations.

The first six items of Table 1 are completely fulfilled by the preclinical EPO studies included in the meta-analysis. However, randomization and blindness are reported only in a few studies whereas diffusion/perfusion MRI or serum markers of tissue injury are not. So far, rhEPO efficacy in gyrencephalic species has not been tested and only one study used hypertensive rats. An EPO–rtPA interaction study in a stroke model was recently published [Zechariah et al. 2010]. All in all, there is still a lot of work to do regarding EPO preclinical studies to fully meet the currently updated STAIR recommendations.
Beneficial effects of both rhEPO and its nonhematopoietic derivatives have also been described in models other than acute ischemic stroke. These molecules improved morphological, functional and cognitive recovery in animal models of traumatic brain injury [Brines et al. 2000; Chen et al. 2007; Mahmood et al. 2007]. rhEPO and its derivatives have also been used in ischemic, traumatic or compressive spinal cord injury models [Gorio et al. 2002; Grasso et al. 2006; Pinzon et al. 2008]. In experimental autoimmune encephalomyelitis (an animal model for multiple sclerosis), EPO and its derivatives improve functional recovery; reduce tissue damage and inflammatory responses [Brines et al. 2000; Leist et al. 2004; Savino et al. 2006]. These drugs have also been successfully investigated in models of peripheral axonal nerve injury, diabetic, and HIV-associated neuropathy [Campana et al. 2006; Keswani et al. 2004; Schmidt et al. 2008]. In these conditions, the anti-apoptotic, anti-oxidative, and trophic effects on both neurons and oligodendrocyte progenitor cells are likely to reduce inflammation and preserve myelination and neuronal function. The latter evidence suggested that EPO or its derivatives would provide a new approach to safety and successfully treat a variety of nervous system disorders.
rhEPO neuroprotective effects in acute ischemic stroke trials
Although preclinical data related to rhEPO and EPO variants have been promising for more than a decade, translation to clinical use has not been expedite. In 2002 the results of the first clinical trial (phase I/II) ‘Gottingen EPO Stroke Study’ were published. Regarding safety, functional recovery and infarct size reduction, these results were reliable and good enough to be considered satisfactory in terms of neuroprotection [Ehrenreich et al. 2002].
However, contradictory information emerged from the ‘Multicenter EPO Stroke Trial’ in 2009, where rhEPO was combined with rtPA. This trial highlighted safety concerns and failed to show any clinical benefit. A large percentage of thrombolyzed patients exhibited unexpected side effects, i.e. brain edema and hemorrhage [Ehrenreich et al. 2009].
Such discrepancies between the previous studies may result from potential rtPA–EPO interactions, not previously studied in preclinical experiments. Recent research in focal cerebral ischemia in mice attributes the negative effect of the rtPA–EPO combination to excessive matrix metalloproteinase activation, promotion of brain edema and DNA fragmentation [Zechariah et al. 2010]. Jia and colleagues also showed that rhEPO exacerbated tPA-induced brain hemorrhage and increased matrix metalloproteinase-9 (MMP-9), NF-kappaB, and interleukin-1 receptor-associated kinase-1 when the combination treatment starts 6 hours after stroke in a rat model of embolic middle cerebral artery occlusion [Jia et al. 2010].
It is noteworthy that the ‘Multicenter EPO Stroke Trial’ did not stick to strict protocol rules in 50% of rtPA-treated patients [Ehrenreich et al. 2009]. Ethical criteria to conduct clinical trials compel researchers to introduce the new approach together with the already approved therapy (rtPA in the present case). However, it is mandatory to undertake preclinical studies to explore possible interactions and administration regimens between the new and the already approved therapy, as every drug combination could be a source of unknown risk. Those studies should be carried out before the clinical trial begins. It is remarkable that in the ‘Multicenter EPO Stroke Trial’, the non-rtPA-treated patients showed beneficial effects similar to those observed in the previous ‘Gottingen EPO Stroke Study’.
Further clinical proofs for rhEPO beneficial effects in ischemia came from studies regarding other brain pathologies such as aneurismal subarachnoid hemorrhage [Tseng et al. 2009] and severe traumatic brain injury [Talving et al. 2010]. A reduction in delayed ischemic deficits as well as an increase of in-hospital survival were observed in the rhEPO treated groups. None of the latter trials reported safety concerns.
The use of rhEPO has also been reported as a feasible and safe treatment for neurocognitive dysfunction, which is a common complication after coronary artery bypass surgery. This procedure is performed in patients with coronary artery disease, the most common cause of death in North America [Haljan et al. 2009]. A trend to reduce neurocognitive dysfunction at 2 months was associated with all EPO doses [Haljan et al. 2009].
In an attempt to define a new restorative intervention for stroke patients, rhEPO has been combined with β-human chorionic gonadotropin (therapy initiated 24–48 h after stroke onset). This three-center, single-dose, phase IIa trial supported the safety of this growth factor therapy [Cramer et al. 2010].
Safety and efficacy studies were undertaken in neonates with hypoxic-ischemic encephalopathy [Elmahdy et al. 2010; Zhu et al. 2009] or extremely low birth weight [Fauchere et al. 2008; He et al. 2008]. In the latter studies rhEPO was well tolerated without any apparent side effects, and the disability risk of infants with mild to moderate hypoxic-ischemic encephalopathy was reduced [Elmahdy et al. 2010; Zhu et al. 2009]. Moreover, low birth weight infants improved their language as well as their developmental gross and fine motor activity quotients [He et al. 2008]. The previous results showed that reliable designs and strict attachment to protocols are strong partners for success.
Conclusions
Brain protection rather than just neuroprotection would be required for stroke therapy, considering that the whole neurovascular unit (endothelium, glial cells, neurons and extracellular matrix) is affected by an ischemic episode. rhEPO therapy remains a good option because of its broad brain effects, which could be potentiated with the use of combined therapies. Nevertheless, any possible interaction between agents to be combined in human therapy should be previously investigated in preclinical studies.
On the other hand, there are still some questions that remain to be answered by new preclinical EPO studies to completely fulfill the STAIR recommendations. For example, as comorbidities are an important issue in determining the outcome of patients after stroke, it is mandatory to perform additional preclinical studies where rhEPO effects would be evaluated in the context of common concomitant diseases. The range of circumstances in which EPO works best should be accurately delineated.
Fortunately, there are several ongoing studies applying rhEPO and its derivatives to treat several human nervous system diseases [Siren et al. 2009] which will surely clarify and reinforce the role of these molecules as neuroprotective agents.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declare no conflicts of interest in preparing this article.