
Abstract
Despite extensive research, treatments for clinical stroke are still limited only to the administration of tissue plasminogen activator and the recent introduction of mechanical thrombectomy, which can be used in only a limited proportion of patients due to time constraints. A plethora of inflammatory events occur during stroke, arising in part due to the body’s immune response to brain injury. Neuroinflammation contributes significantly to neuronal cell death and the development of functional impairment and death in stroke patients. Therefore, elucidating the molecular and cellular mechanisms underlying inflammatory damage following stroke injury will be essential for the development of useful therapies. Research findings increasingly point to the likelihood that epigenetic mechanisms play a role in the pathophysiology of stroke. Epigenetics involves the differential regulation of gene expression, including those involved in brain inflammation and remodelling after stroke. Hence, it is conceivable that epigenetic mechanisms may contribute to differential interindividual vulnerability and injury responses to cerebral ischaemia. In this review, we summarize recent findings on the emerging role of epigenetics in the regulation of neuroinflammation in stroke. We also discuss potential epigenetic targets that may be assessed for the development of stroke therapies.
Introduction
An ischaemic stroke is due to a blockage of an artery supplying blood to the brain, resulting in cerebral infarction with accompanying symptoms such as sudden weakness, facial numbness, disability in speech and sight, or paralysis. 1 A blockage can be caused by a blood clot formed within the artery (thrombotic stroke) or may be formed elsewhere, such as in the heart (known as an embolus), which then travels via the arterial system to the brain, causing embolic stroke. Stroke aetiology differentiation is also related to large or small vessel disease, whereas embolic strokes can be differentiated into cardiac embolic strokes or strokes with an arterial source (artery to artery). 2 The formation of blood clots within an artery may be related to the presence of atherosclerosis,1,3,4 diabetes mellitus, coronary heart disease, hypertension, 5 as well as hyperlipidaemia. 6 During an ischaemic stroke, a pathway of events known as the ‘ischaemic cascade’ is activated temporally and spatially and is responsible for damage in the affected cerebrovascular tissue. In the ischaemic cascade, events such as energy failure, peri-infarct depolarization, excitotoxicity, oxidative stress, and inflammation work in concert to cause rapid cell death in the affected tissue.1,7–9
During a cerebral arterial occlusion, the presence of a thrombus results in stagnant blood flow, which triggers a series of inflammatory cascades. 4 The neuroinflammation process may also be triggered during reperfusion, resulting in further damage to the brain. Current understanding of stroke-induced inflammatory mechanisms is reviewed elsewhere by us and others.1,4,10–15 Briefly, the inflammatory cascade is initiated via the molecular release of cytokines and chemokines by inflammatory cells within the ischaemic territory, which leads to the activation of endothelial cells to upregulate numerous inflammatory mediators that facilitate leukocyte infiltration into the brain parenchymal region. Infiltrated leukocytes produce and release cytotoxic and proinflammatory chemicals that induce toxicity to neurons and glial cells. In addition, activation of the inflammasome complex in various brain cells leads to the production of proinflammatory cytokines such as IL-1β and IL-18.10,16,17 Similarly, the complement cascade is activated in neuronal and glial cells. 18 Collectively, these mechanisms lead to structural and functional impairment of neuronal cells in the ischaemic area.
Many factors have been identified that affect stroke risk and functional outcome. Risk factors for stroke are numerous, and include lifestyle factors such as obesity, diabetes, smoking, advanced age and lack of physical activity.19,20 Thus, as the pathogenesis of stroke is known to be impacted by such environmental/external factors, there opens up a wider area of interest as to whether stroke incidence and outcome might also be influenced by epigenetics.
Gene expression can be modulated via changes in the DNA sequence itself, which may even be heritable if changes occur in DNA sequences affecting germ cells. ‘Epigenetics’ refers to the interaction of environmental factors with the genome that may also result in heritable and modifiable gene expression or phenotype, which does not confer any changes in the DNA sequence itself.21,22 The eukaryotic genome is tightly regulated in terms of its organization and differential control mechanisms from the DNA sequence to the post-translational level. At every level of eukaryotic control, such regulatory processes are being controlled by another layer of epigenetic regulation. As such, exposure to various environmental stimuli may alter the epigenome status, which in turn differentially controls the modulation of gene expression and protein activity. As such, higher-order DNA activity is modulated by a dynamic interaction between genes and environmental factors. Epigenetic processes thus serve as an important spatial and temporal regulator of a number of biological processes in the body, such as homeostasis, development and ageing.23,24
Recently, much attention has been shifted towards the study of epigenetics in influencing the risks and manifestation of various diseases, such as cancer. Epigenetic markers represent a useful and reliable prognostic risk biomarker and can be used to explain individual susceptibility towards the pathogenesis of diseases. However, stroke is not manifested as a monogenic disease, but represents a complicated polygenic disease that especially affects the ageing population, and is often confounded by many lifestyle-related metabolic disorders. As individuals are subjected to a myriad of environmental factors throughout life, it is possible that stroke incidence and outcome may be differentially regulated by epigenetic mechanisms between individuals. This may help to explain why outcomes from studies conducted on rodent models may be poorly translated into human stroke patients as their epigenomes will differ greatly. Until recently, epigenetic studies in stroke have been in their infancy, and relevant information is only just beginning to emerge.
While conventional therapeutic approaches that aimed to intervene against ischaemic stroke damage have been unsuccessful, new approaches have started to shift attention towards the area of regenerative medicine. Regenerative therapeutic approaches now aim to attenuate inflammatory-induced damage, to promote neuroprotection and neural repair, prevent ischaemic-induced cell death, as well as maintain structural and functional homeostasis by promoting cerebral remodelling and regeneration. These regenerative processes seem to be tightly regulated via epigenetic mechanisms, and depending on the state of the epigenome of the individual, the degree of regeneration may differ and so the extent of damage incurred will vary. As such, it is paramount that studies consider the complex and interrelated genetic and molecular interactions from an epigenetic viewpoint. This review mainly focuses on how epigenetic mechanisms might contribute to post-ischaemic neuroinflammation and neuronal cell death. The next section will describe mechanisms of epigenetics and epigenetic regulation of the inflammatory process that contributes to infarct development following ischaemic stroke.
Epigenetic mechanisms
The eukaryotic genome has a distinct organizational structure interlaced with multilayer packaging and folding. Chromosomal DNA is often associated with histone proteins to form a structural core, termed the nucleosome. Each nucleosome consists of a 147-base-pair DNA wrapped around an octamer of histones made up of a pair of H2A and H2B dimers, as well as a pair of H3 and H4 dimers. Between two nucleosomes, DNA not associated with nucleosomes (coined ‘linker DNA’) often associates with H1 histone proteins. This interaction is possible as DNA is negatively charged and is able to form tight binding with positively charged histone proteins (rich in basic lysine and arginine residues).25,26 As DNA replication and transcription during gene expression is dependent on the accessibility of replication and transcriptional machinery to DNA sites, nucleosomes thus serve as a form of steric hindrance that impede the access of such machinery. 27 The relative packing of DNA with nucleosomes will determine the overall accessibility of DNA, defining two main regions termed ‘euchromatin’ and ‘heterochromatin’. Euchromatin is defined as chromatin that is less tightly packed and highly involved in transcription, whereas heterochromatin represents the opposite. The dynamic transition between a euchromatin and heterochromatin state is highly dependent on epigenetic modifications that occur on the DNA sequences or on amino histone tails. 28 In this review, for didactic purposes, these epigenetic modifications and mechanisms can be broadly categorized into DNA methylation, histone modifications and microRNA involvement. The overall epigenetic tags that are imprinted across the genome is termed the ‘epigenome’, which can be identified as microdomains residing within the nucleus, which in turn will regulate the overall DNA structure and accessibility to provide differential patterns of gene expression. 29 In simpler terms, the overall epigenome can be viewed as a tug-of-war, in which different epigenetic tags will either regulate positive or negative gene expression, and the sum of this dynamic interaction will determine the direction of gene expression.
DNA methylation
At the DNA sequence level, DNA methylation is one of the most well-studied epigenetic modifications. An epigenetic tag involves the covalent attachment of a methyl group to a cytosine ring at carbon position 5 (Figure 1). While cytosine is one of the principal conserved residues present throughout DNA, DNA methylation does not occur on every cytosine residue in the genome. Rather, the process of DNA methylation is biased, occurring only in regions termed CpG dinucleotide islands. These islands are recognized by stretching over more than 500 base pairs of DNA, with guanine and cytosine composition to be above 55% frequency, as well as an overall observed frequency ratio of CG:GC to be at least 0.6. 24 These CpG islands are normally located in the promoter region of genes, which also tends to make up the 5′ gene transcript. Besides that, it has been recently identified that while CpG islands exist throughout the genome, their relative density differs across different positions. 27 Another term, coined the ‘CpG shores’, 2kb downstream of Cpg Islands, there exists a much lower density of Cpg sites termed Cpg shores. These Cpg shores contain DNA methylation sites, which challenges conventional norm that DNA methylation only occurs at high density CPg sites. 24 As such, DNA methylation does not only occur at high-density CpG islands, but also at locations that contain low-density CpG residues, which challenge conventional understanding of this relatively important and well-known epigenetic tag mechanism.
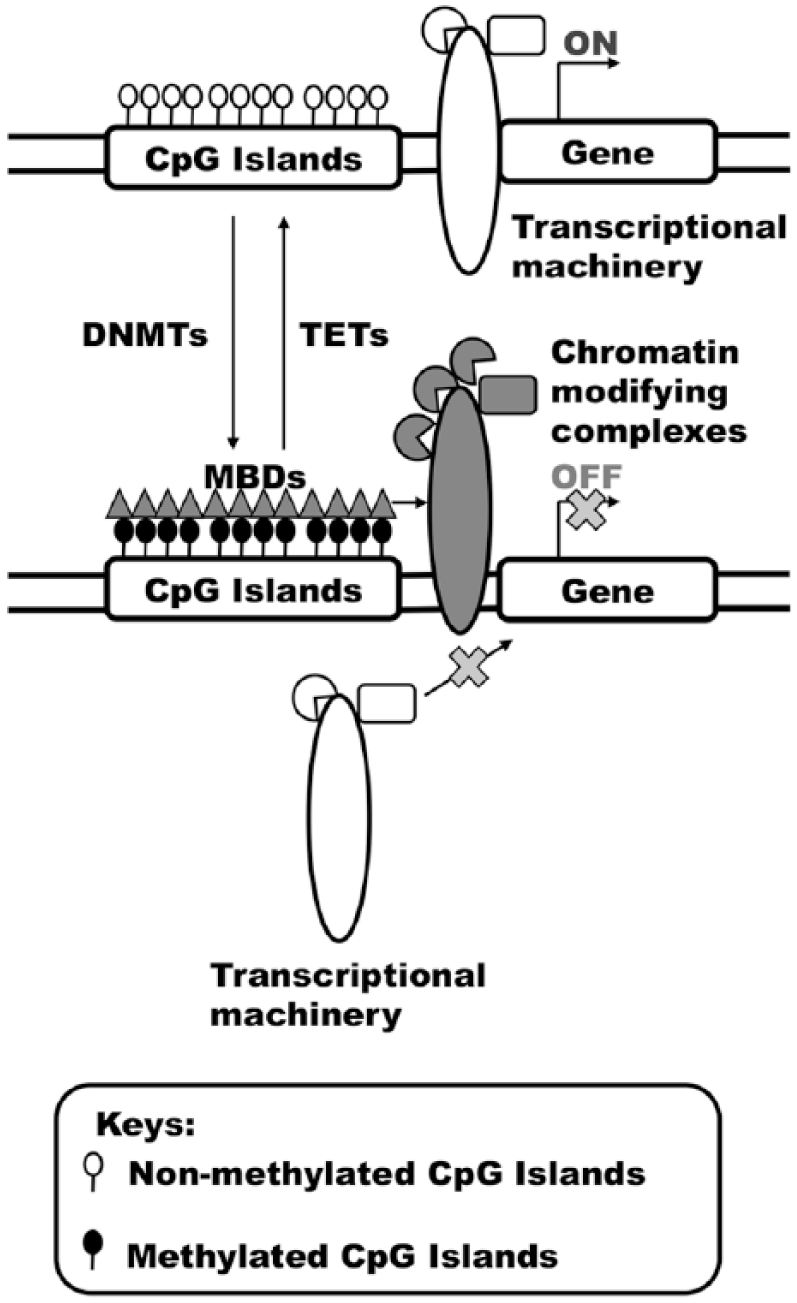
DNA methylation. DNA methylation sites displayed bias at CpG islands or shores. Hypomethylation of CpG islands is normally associated with transcriptional activation. DNA methylation is mediated by a group of enzymes termed DNA methyltransferases (DNMTs), whereas the subsequent removal of methyl group from DNA is mediated by ten–eleven translocation (TET) enzymes. Methyl tags at CpG islands attract methyl-CpG-binding domain (MBD) proteins, which may recruit chromatin-modifying complexes to creates a repressive chromatin, leading to gene silencing.
DNA methylation is often associated with transcriptional inactivation. The addition of a methyl group to CpG sites can prevent gene transcription via various mechanisms. DNA methylation can directly prevent the binding of DNA-binding factors to transcriptional sites. Moreover, the addition of methyl groups to CpG sites can be recognized by a family of proteins termed ‘methyl-CpG-binding domain (MBD) proteins’. Binding of these proteins to methylated CpG sites will in turn recruit histone or chromatin-modifying complexes, which in turn will assemble a spectrum of complexes that centrally mediate the repression of gene transcription. The addition of methyl groups to CpG sites is mediated by DNA methyltransferase (DNMT) family member enzymes. 30 Of the five family members reported within this family of enzymes, only DNMT1, DNMT3a and DNMT3b possess enzymatic activity. By transferring a methyl group from S-adenosyl methionine to DNA, DNMT3a and DNMT3b are primarily involved in de novo methyl group transfer, whereas DNMT1 is involved in the maintenance of DNA methylation status in the epigenome.31,32 The cellular reservoir of S-adenosyl methionine is maintained by another group of enzymes called ‘methylenetetrahydrofolate reductases’ (MTHFRs). This enzyme group plays a critical role in the metabolism of folate and methionine, which are needed to support the role of DNA synthesis or methylation.33–35 On the other hand, recent findings have discovered the presence of TET enzymes, which help to mediate DNA demethylation via a sequential process. 36
Surprisingly, while DNA methylation represents a prevalent form of epigenetic modification, the presence of DNA methylation in the mammalian genome is relatively rare (~1%). This is because 5-methylcytosine is genetically unstable in DNA sequences as it cannot be excised or repaired by the DNA repair system. As a result, 5-methylcytosine undergoes spontaneous deamination to thymine, otherwise the presence of this methylated residue will be a potential mutational hotspot. The consequence of this is the rapid depletion of CpG sites in the genome, where the remaining CpG sites are subsequently scattered around the genome in low proportions. 37 Despite the low proportion of CpG sites within the genome, DNA methylation is still critical in many biological processes besides gene silencing, such as differentiation, genomic imprinting, genomic stability and X chromosome inactivation.
Histone modifications
Histones that form part of the nucleosome structure represent a globular structure with their amino terminal tail protruding from each subunit. These histone tails are subjected to a myriad of post-translational modifications, such as methylation, acetylation and phosphorylation. These post-translational modifications of histone tails play various critical functions, regulating important aspects of chromatin packing and organization, transcriptional activation or repression, DNA repair, as well as telomere maintenance. Compared to DNA methylation, histone modification represents a relatively short-term reversible change that is especially sensitive to external stimuli changes.38–40 As different histone modifications confer different cellular outcomes, the distinct expression of total histone modifications, termed the ‘histone code’, will in turn determine the sum of the total interacting changes in response to a particular stimulus to produce the overall cellular outcomes.
Acetylation of histones
Acetylation of histone tails has been widely investigated for its role in influencing chromatin accessibility as well as gene expression. The covalent addition of acetyl groups to histone tails at the epsilon-amino group of conserved lysine residues is regulated by a family of writer enzymes termed histone acetyltransferases (HATs) (Figure 2). It has been generally described that histone acetylation is associated with a permissive chromatin state and drives transcriptional activation. 41 This may be achieved via the synergistic interaction with nucleosome remodelling complexes, such as the SWI/SNF-lie ATPase family members with HATs.42–44 Nucleosome remodelling may result in the sliding of nucleosome away from DNA through weakening of DNA–histone interaction, thereby promoting the accessibility of DNA to transcriptional machineries. On the other hand, the removal of acetyl groups is regulated by eraser enzymes known as histone deacetylases (HDACs).45,46 Deacetylation of histone tail residues may also recruit repressive proteins and thereby result in a repressive chromatin state that generally silences transcription. 41 As histone acetylation and deacetylation is a highly volatile process that is extremely sensitive to stimulus changes, their regulatory influences on the chromatin structure and subsequent functional importance in gene expression has drawn much attention in the investigation of epigenetic and disease progression. Besides that, other significant processes that are regulated by histone acetylation or deacetylation include transcriptional elongation and DNA repair.46–49

Histone acetylation. Histone acetylation occurs at the amino termini of histone tails, and is mediated by a class of enzymes termed histone acetyltransferase (HATs). Subsequent removal of an acetyl group is mediated by another class of enzyme called histone deacetylases (HDACs). Histone acetylation is commonly associated with permissive chromatin accessibility and transcriptional activation.
Methylation of histones
Histone methylation represents another form of histone modification that also helps to regulate chromatin accessibility and the level of gene expression (Figure 3). Compared to DNA methylation, methylation at histone levels occurs at the amino terminal tails, and the effects on gene expression are often reversible and complex. The addition of methyl groups onto the histone amino terminal tails normally occurs at either lysine or arginine residues.26,50 However, given the conservative nature of residues that are able to be methylated, it has been generally observed that histone methylation occurs at different positions, and the effects generated are often dependent on the positions of the residues being methylated. Besides that, at a specific residue, the addition of methyl groups can be mono-, di- or trimethylated at lysine residues, whereas in the case of arginine residues it can either be mono- or dimethylated.26,51 Dimethylated arginine residues have another layer of complexity, where the methylation can either be in a symmetrical or asymmetrical topology.50,52 As a result, given the possible permutations and combinations of methylation patterns, it is therefore difficult to predict the effects of histone methylation on the transcription of genes. Despite the complexity of the mechanisms governing histone methylation, it has been widely reported that certain well-known methylation marks have been associated with transcriptional activation and repression.29,51,53 For instance, methylation at histone 3 lysine 4 is generally associated with transcriptional activation, whereas methylation at histone 3 lysine 9 is transcriptional repressive. 54 Furthermore, the reversible nature of histone methylation and demethylation is mediated by the dynamic interaction between lysine or arginine methylases or demethyltransferases. The coordinated action and interplay between these readers and erasers will lead to diverse methylation marks, which in turn will regulate gene expression spatially and temporally.

Histone methylation. Histone methylation normally shows preference at either lysine or arginine residues of histone tails. Addition of methyl groups to these residues is mediated by either lysine or arginine methyltransferases, respectively. Methyl group addition to arginine residues can either be symmetrical or asymmetrical, and is mediated by several subfamily members of arginine methyltransferases. Unlike histone acetylation, effects of histone methylation on gene transcription are still unclear. Both transcriptional activation and deactivation have been reported to be associated with histone methylation.
MicroRNAs
MicroRNAs (miRNAs) represent an important emerging player in the field of epigenetics, involving precise interaction with specific genes and the ability to modulate the level of gene expression. miRNAs belong to a class of short non-coding RNAs, which exert their action at the post-transcriptional level, and are an important fine-tuner in the control of gene expression. 39 Biogenesis of miRNAs is often complicated and involves multi-sequential steps (Figure 4). miRNA genes are first transcribed in the nucleus by RNA polymerase II into a primary transcript. This primary transcript, termed primary miRNAs (pri-miRNAs), is often very large and contains multiple miRNA genes occupying various loci across the pri-miRNAs. Following this, these pri-miRNAs will undergo micro-processing by a class of RNaseIII enzymes called a Drosha/DGCR8 complex. 55 This process will result in the generation of shorter hairpin–loop structures, called ‘precursor miRNAs’ (pre-miRNAs). Subsequently, pre-miRNAs will be exported out of the nucleus into the cytoplasm via the action of Exportin-5 protein. 56 In the cytoplasm, pre-miRNAs will undergo a second round of cleavage by another RNaseIII enzyme termed ‘Dicer’, which will produce a duplex of the mature miRNAs. 57 To mediate its biological activity of gene silencing, this duplex of mature miRNAs will undergo asymmetrical unwinding by the Dicer/TRBP complex, where a single strand of the mature miRNAs will then associate with a ribonuclear particle to form the RNA-induced silencing complex (RISC). The mature miRNAs in the RISC complex have been found to commonly recognize the 3′ untranslated region (3′ UTR) of the mRNAs, which may suppress protein synthesis or mediate mRNA degradation. 58 miRNAs play many important biological roles in organisms, controlling the processes of cellular proliferation, differentiation and metabolism, as well as development. 59 Recently, miRNAs have been reported to be dysregulated in many diseases, such as cancer and neurodegenerative diseases. Interestingly, miRNAs have also been recently proposed to be a useful and accurate disease biomarker for diagnosis and prognosis prediction. However, given that miRNAs can only recognize partial sequences in mRNAs, miRNAs can recognize a myriad of mRNAs simultaneously and work differently in a cell-context-dependent manner. As such, studies investigating the roles of miRNAs in diseases need to understand the complicated regulatory network that miRNAs operate. 60

MicroRNAs. miRNA processing follows a complicated cascade. Transcription in the nucleus first produces a long transcript containing numerous miRNA transcripts by RNA polymerase II, termed primary miRNAs (pri-miRNAs). Subsequent processing by Drosha/DGCR8 complexes yields a precursor miRNA (pre-miRNA) to be transported via Exportin-5 towards the cytosol. In the cytosol, pre-miRNAs undergo a second round of processing to become mature miRNAs via interaction with the Dicer/TRBP complex. Eventually, these mature miRNAs will be packaged into an RNA-induced silencing complex (RISC). Together with RISC, mature miRNAs tend to recognize the 3′ untranslated region (3′ UTR) sites of messenger RNA (mRNA) specifically. Targeting by mature miRNAs will either lead to mRNA silencing or degradation.
Epigenetic regulation of inflammation in stroke
DNA methylation and inflammation in stroke
Using in vitro experiments, an ischaemic insult to cultured cells results in hypomethylation in the global genomic landscape. However, depending on the temporal aspect of stroke progression, certain regions of the genome experience enhanced DNA methylation. These results may reveal a temporal regulation of DNA methylation in response to stroke, which may play different roles in neurotoxicity and neuroprotection. 61 Besides that, studies have shown that a low level of methylation in blood LINE-1 repetitive sequences is associated with higher stroke risk and poorer prognosis and mortality. 62 Recent studies have reported that hypomethylation of LINE-1 DNA sequence is associated with higher VCAM-1 levels, which may promote stroke-induced inflammation and thereby exacerbate stroke injury. 63 Hypomethylation of CpG sites of TNF receptor associated factor 3 (TRAF3) is also associated with enhanced platelet conglomeration in patients receiving clopidogrel (an antiplatelet drug), thus increasing the recurrence of ischaemic stroke. 64 Conversely, hypermethylation of the thrombospondin-1 (THBS1) gene promotor region leads to gene silencing. THBS1 is secreted by platelets and is an inflammatory mediator needed to induce angiogenesis during cerebral ischaemia, which leads to neurorepair.65,66 Gene silencing of THBS1 via DNA methylation during stroke prevents neurorepair, thus exacerbating stroke injury.58,67,68 Similarly, another study has shown that methylation of protein phosphatase magnesium dependent 1A (PPM1A), which is involved in the inflammatory healing process, have led to increased risks of vascular recurrence in stroke patients who are treated with aspirin. 69 As such, epigenetic modulation of DNA methylation during stroke may regulate inflammatory damage and repair processes during stroke injury, as well as subject patients to chronic inflammation that further increases stroke recurrence. However, other studies have reported contradictory results that no significant changes were observed in global DNA methylation status in large-artery atherosclerosis stroke, small-artery disease stroke, as well cardio-aortic embolism stroke. 70 As such, DNA methylation may be confounded by different pathologies. Careful consideration and further studies are needed to gain a deeper understanding of the roles of DNA methylation in different stroke subtypes. Nonetheless, it has been recently proposed that the level of DNA methylation correlates with the chronological age of patients and serves as a good prognostic marker to assess stroke risk. 71
It has been reported that DNMT3a is responsible for degeneration of motor neurons in vitro using cultured NSC34 cells. Moreover, both DNMT1 and 3a are upregulated during induced apoptosis of NSC34 cells. 72 Using the DNMT inhibitor RG108 in vivo, methylation of CpG sites is also inhibited with the concomitant blockage of apoptosis of motor neurons. 72 However, this study is being conducted with the amyotrophic lateral sclerosis mechanism in mind. Moving forward, it will be interesting to investigate the roles of DNMT in neuronal cell death or glial cell activation following ischaemic stroke. DNA methylation is also reported to regulate differentiation and maturation of various cell types. 30 Post-mitotic neurons are highly enriched in DNMT1 and DNMT3a. 73 While studies have linked these enzymes to synaptic plasticity in learning and memory, 31 further studies are required to investigate the roles of these enzymes in the neuronal response in ischaemic stroke. Furthermore, it has been established that enhanced expression of DNMT1 in macrophages leads to downregulation of peroxisome-proliferator activator receptor gamma (PPARγ), which helps to upregulate the expression of proinflammatory cytokines that drives atherosclerosis in mice. 74 Another study also reported that DNA methylation profiles represent a useful biomarker to assess atherosclerotic progression in the human aorta. 75 This lends useful insight on the roles of DNA methylation in association with atherosclerosis pathogenesis, which is a major risk factor for ischaemic stroke. In a mouse model of ischaemic stroke, mild ischaemic insult resulted in upregulation of DNA methylation, which is correlated with poor stroke outcome. 76 Inhibition of DNMT resulted in neuroprotection from this mild ischaemic damage. However, using another stroke model that encapsulates excitotoxicity and necrotic death, DNMT is not involved in stroke outcome. 76 These findings may highlight the potential roles of DNMT in mediating pathological outcomes in response to various degrees of ischaemia, and also the importance of variability in studying epigenetics using different stroke models. Furthermore, DNMT1 has been discovered to regulate the crosstalk between major immune signalling pathways, such as T-cell receptor (TCR) and B-cell receptor (BCR) pathways. 77 DNMT1 downregulates the activity of TCR/BCR pathways, which drives immunosuppression in cardiovascular disease and stroke, 77 which may explain the higher risks of infection associated with post-stroke patients. 78
In addition, methylenetetrahydrofolate reductase (MHTFR) has also been implicated to trigger inflammatory responses and thereby is correlated with increased stroke risk. 79 MHTFR is an enzyme responsible for the catalytic regeneration of methionine, which eventually is required as a methyl donor for DNA methylation. 80 For many years, it has been widely investigated that individuals exhibiting polymorphism of C677 > T demonstrate hyperhomocysteinemia, a condition characterized by accumulation of homocysteine, a product formed following DNA methylation. 80 MHTFR is responsible for the reconversion of homocysteine to methionine. 81 However, individuals possessing this genetic variant lack the ability to regenerate the methyl reservoir, leading to the build-up of homocysteine. 82 Interestingly, individuals possessing this genetic variant often have increased risk of cardiovascular and stroke events. 83 Elevation in the homocysteine level is able to induce a plethora of inflammatory responses within the cerebral territory. Within endothelial cells of the vasculature, high levels of homocysteine are able to induce oxidative stress, as well as generate a myriad of proinflammatory mediators such as TNF and inducible nitric oxide synthase (iNOS). 84 Together, hyperhomocysteinemia is able to drive the pathogenesis of endothelial dysfunction, which promotes the development of cerebral vascular damage and increases the risk of stroke.33,35,85–87 However, in other studies, MHTFR polymorphism association with stroke risk is contradictory. In one study conducted on the Black Sea Turkish population, MHTFR polymorphism did not seem to influence the risk of ischaemic stroke. 88 Moreover, in a North Indian population, MHTFR polymorphism was also not correlated with ischaemic or haemorrhagic stroke risk. 89 Besides that, this observation is consistent with results reported in Central and Northern Europe, whereas in Italy, MHTFR polymorphism is associated with ischaemic stroke risk. 34 The presence of heterogeneous observations with regards to MHTFR polymorphism and stroke risk may be due to confounding factors like ethnicity, as well as inconsistency in the sample size within the populations studied. Interestingly, while genetic variants may account for the manifestation of hyperhomocysteinemia, it has also been discovered that the level of homocysteine within the body is epigenetically regulated. In one study, a young patient with a reported case of concomitant Crohn’s disease and C677 > T polymorphism displayed increased inflammation, as well as deficiency in vitamin B6 levels. 90 This patient subsequently developed a large-artery stroke, highlighting that the increased risk of stroke may be confounded by other factors. 90
Many factors have since been identified to modify the serum level of homocysteine, including lifestyle factors (e.g. age, smoking, diet and drugs). 82 For instance, low vitamin (e.g. B6 and B12) and folate levels and migraine have been associated with increased risk of stroke.91–93 As such, many studies have started to investigate whether modifying these environmental factors might also modify the plasma level of homocysteine, and thus provide an epigenetic approach to attenuate stroke risk. It has been reported that an appropriate intake of vitamins, antioxidants, as well as folic acid supplementation, confers protection against stroke.91,94,95 However, one group reported that DNA methylation of the MHTFR gene that mediates vitamin B12 and serum folate levels increased the risk of ischaemic stroke. 96 As such, it is still unclear whether the association of MHTFR with stroke risk is due to the deficiency of these dietary cofactors or the epigenetic regulation of MHTFR that leads to these deficiencies in the serum. More studies need to be undertaken, but clinical trials have started with these dietary cofactors in stroke prevention.91,95
Histone acetylation and deacetylation in stroke
Using in vitro studies on murine and rat neuroglial cells, it has been shown that the administration of HDAC inhibitor trichostatin A helps to drive proinflammatory responses such as the upregulation of IL-6, iNOS and TNF. 97 This proinflammatory response is attenuated via inhibition of NF-κB, which suggests that the modulation of histone acetylation status in microglial cells could act through an NF-κB-dependent manner to drive the proinflammatory response. 97 Another group has also reported that acetylation at histone 3 lysine 9 (H3K9Ac) is upregulated in the ischaemic territory and is associated with microglial activation. 98 Inhibition of HDAC in an in vitro model found that H3K9Ac upregulation leads to decreased expression of proinflammatory genes like TNF, IL-6, iNOS and STAT1, as well as an increase in anti-inflammatory genes like IL-10 and STAT3. 99 The overall effect improved neuronal survival consistent with a neuroprotective action of HDAC inhibitors. 99 Notably, it has been reported that trichostatin A-mediated neuroprotection via HDAC inhibition was not present in gelsolin-deficient mice after ischaemic stroke. 100 It was also demonstrated that trichostatin A-mediated HDAC inhibition was highly dependent upon the expression of gelsolin, in addition to NF-κB.97,100 Furthermore, in a rat model of intracerebral haemorrhage, administration of the anti-epileptic drug valproic acid (VA) reduces inflammation and brain injury. Such changes were mediated via an interaction of VA with HDAC, leading to its inhibition. 101 Moreover, in a rat model of focal cerebral ischaemia, VA administration was reported to ameliorate blood–brain barrier disruption and subsequent brain oedema through the downregulation of matrix metalloproteinase 9 (MMP-9), tight junction degradation, and the NF-κB pathway via suppression of HDAC. 102 Moreover, another HDAC inhibitor, sodium butyrate, is reported to downregulate proinflammatory mediators IL-1β and IL-18, and increase expression of the neuroprotective protein insulin growth factor 1 (IGF-1) in ischaemic stroke. 103 In a rat model of permanent ischaemic stroke, the post-stroke injection of HDAC inhibitors, sodium butyrate, trichostatin A or VA, improved functional outcome. 104 This amelioration of stroke-induced tissue injury was mediated through inhibition of H3 histone deacetylation, which in turn reduced inflammation as assessed by microglia number and activity, and expression of proinflammatory markers. 104 HDAC inhibitors also upregulated expression of heat shock protein 70 (Hsp70), a neuroprotective protein, and reduced expression of phosphorylated AKT, p53, iNOS and cyclooxygenase-2, in the ischaemic territory. 104 Importantly, these inhibitors conferred protective benefits when administered at around 3 h after stroke onset, which is a potentially relevant therapeutic window for use in humans.
It has been established that ischaemic stroke causes a drastic reduction in H3 acetylation. 105 Treatment with another HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA), prevents this reduction in neurons and astrocytes and improves stroke outcome. 105 Consistent with the previous study, SAHA treatment resulted in upregulation of Hsp70 and the anti-apoptotic Bcl-2, as well as reduced expression of proinflammatory markers such as IL-1.105,106 Similarly, it has been reported that stroke induces a drastic decrease in histone 3 lysine 9 lysine 14 acetylation (H3K9K14Ac), 107 with a myriad of genes also dysregulated, particularly those associated with HDAC3. 107 Again, administration of the HDAC inhibitor, SAHA, reduces the level of neuroinflammation and ischaemic cell death. 107
It is important to note that the epigenetic status of one cell type in the ischaemic territory may also influence the activity of neighbouring cells. Microglial activation can influence the reduction in acetylation of H3 and H4 histones in astrocytes, thereby leading to the downregulation of astroglial nuclear factor-erythroid 2-related factor 2 (Nrf2), a mediator important in anti-oxidant defence in astrocytes. 108 Treatment with the HDAC inhibitor VA or the GSK3β inhibitor lithium can restore the level of acetylation and thereby maintain anti-oxidant defence previously suppressed due to inflammation. 108
It is important to note that the HDAC family described in the studies above consists of many members. While HDAC inhibitors have been shown to be protective against stroke-induced damage, most studies using HDAC inhibitors did not investigate drug specificity against individual HDAC members. In a study of focal cerebral ischaemia induced by photothrombosis, HDAC inhibition using trichostatin A was ineffective in HDAC2 knockout mice. 109 Functional improvement in stroke outcome was observed when HDAC2 was present, and inhibition of HDAC2 using SAHA improved functional recovery. 109 This demonstrates the importance of considering the roles of individual HDAC members in stroke pathogenesis. Moreover, using RNA interference, another group found in overexpression and mutant studies that HDAC4 forms a complex with nuclear hormone receptor corepressor (N-CoR). 110 Together, this complex is then able to induce the recruitment of monocyte enhancer factor 2 (MEF2) transcription to regulate IL-2 expression. 110 Likewise, HDAC11 regulates expression of IL-10 in antigen-presenting cells. 111 HDAC3 has been reported to contribute to neurotoxicity-induced death of neurons, such that inhibiting HDAC3 with IGF-1, or inhibiting GSK3β, ameliorates this neurotoxicity. 112 Thus, different HDAC members appear to operate differentially across cell types and via varying mechanisms of action. As such, understanding how specific members of HDAC regulate expression of inflammatory mediators will be critical to inform the development of drugs to target epigenetic mechanisms in stroke. Overall, it appears that inhibition of HDAC during cerebral ischaemia confers neuroprotective effects via multiple mechanisms.
The concepts of ischaemic preconditioning and the induction of ischaemic tolerance have recently gained increased interest. Briefly, it has been hypothesized that an intentional ischaemic insult to the brain will induce a plethora of genetic reprogramming resulting in a state of tolerance that will limit the pathogenesis following a subsequent stroke. Such changes in gene expression are thought to confer a neuroprotective effect to reduce the risk and damage associated with stroke. 113 Regarding epigenetics, ischaemic preconditioning has been reported to induce a pro-acetylation status in H3 and H4 histones, which is neuroprotective during cerebral ischaemia. Many strategies have been utilized to induce ischaemic preconditioning. One example of induction of ischaemic tolerance is the chronic dietary intake of acetate. 114 It has been reported that acetate supplementation promoted the acetylation of H3K9, H4K8 and H4K16. 114 Acetylation at these sites has been shown to reduce neuroglial inflammatory responses, as well as the secretion of proinflammatory cytokines like IL-1β. 114 Besides this method, other approaches have been investigated and are reviewed elsewhere. 115
The level of histone acetylation during stroke is also an important determinant for functional recovery. Stroke patients may suffer from memory and cognitive impairment, 116 and HDAC2 has been reported to be a negative regulator of learning and memory, as well as of synaptic plasticity. 117 Treatment with HDAC inhibitors activates the downstream cAMP response element binding protein (CREB): CREB-binding protein (CBP) complex, which promotes memory formation. 118 In rats subjected to stroke, administration of apigenin inhibits HDAC and promotes upregulation of memory formation mediators such as CREB and brain-derived neurotrophic factor (BDNF). 119 Further, cognitive impairment in post-stroke dementia (PSD) – another common pathology in stroke patients 120 – can be ameliorated by treatment with the flavonoid icariin, which promotes histone acetylation in mice. 120 Thus, while HDAC may be an important mediator of inflammatory responses during stroke, targeting HDAC may attenuate inflammatory damage and promote functional outcome. In contrast, HDAC4 has been reported to be a positive regulator of learning and memory formation. 121 Thus, specific members of the HDAC family appear to operate in a differential manner, and careful consideration needs to be given when targeting HDAC members in order to avoid toxicity.
Histone methylation and demethylation in stroke
Stroke outcome is known to be variable in adult and middle-aged rodent models, 54 in association with functional impairment of astrocytes with increasing age that may be related to histone modifications. Following ischaemic stroke, histone 3 lysine 4 trimethylation (H3K4me3) is more highly enriched in astrocytes of young adults than is histone 3 lysine 9 trimethylation (H3K9me3), unlike in middle-aged rats. 54 Histone methylation in a site-specific pattern results in more highly packed chromatin, leading to modulation of vascular endothelial growth factor and microRNA-20 expression. 54 Besides providing insight as to how age may affect the severity of stroke, histone methylation at different sites may also differentially affect downstream gene expression. As such, investigation into the relative expression of histone methylation tags in stroke may allow us to decipher more detail about the molecular mechanisms governing the pathogenesis of stroke.
Using an in vitro neuronal model of stroke, inhibition of H3K9 methyltransferase enzymes, G9a and SUV39H1, using chaetocin and RNA interference demonstrated neuronal resistance to cell death, potentially mediated by more active transcription of BDNF at its promoter site. 122 Furthermore, during rat transient global cerebral ischaemia in vivo, levels of lysine-specific demethylase 1 (LSD-1) are reported to correlate with H3K4 mono-, di- and trimethylation levels in the brain. 123 Interestingly, LSD-1 expression differs spatially and temporally following cerebral ischaemia, which may highlight the particular vulnerability of specific areas of the brain to stroke injury.21,123 Moreover, mild to moderate ischaemia perturbs the activity of histone lysine methylases (KMTs) and histone lysine demethylases (KDMs), thereby leading to a global reduction in histone 3 lysine 9 dimethylation (H3K9me2) in the striatum. 50 Treatment with dimethyloxalylglycine (DMOG), an inhibitor of KDM4 or JMJD2 types of histone lysine demethylases, increases expression of H3K9me2 with concomitant improvement in neurological function after stroke. 50 Modulation of histone methylation levels may thus be a potential target to regulate stroke-induced damage or recovery. Furthermore, many studies also demonstrate a link between histone lysine methylases and demethylases in inflammation. TNF is reported to reduce methylation of H3K9 and H3K27, which is involved in the upregulation of ICAM-1 in cerebral vessels. 124 Using both inhibitor and overexpression studies, it has been shown that G9a lysine methylases and KDM4B lysine demethylases are implicated in modulating the level of methylation at these two sites, which mediates the influence of TNF on expression of either ICAM-1 or VCAM-1, thereby affecting neutrophil infiltration. 124 Notably, in human stroke patients, the levels of serum TNF are reported to be correlated with H3K9Ac and H3K4me3, which may influence stroke outcome. 125 Clearly, epigenetic mechanisms represent complex processes that may be impacted by other histone modifications. HDAC inhibition has been reported to increase H3K4me2 at the Hsp70 promoter region in neurons and astrocytes. 126 As such, epigenetic modulation of a specific histone mark may not be sufficient to induce a change, but rather it may be more useful to consider interactions relevant to the status of the entire epigenome.
Besides histone lysine methylation, histone arginine may also play an inflammatory role that exacerbates stroke injury. It has been reported that rats with hyperhomocysteinemia have relatively low levels of asymmetric monomethylarginine (ADMA). 53 As hyperhomocysteinemia is associated with increased inflammation and stroke risk, findings of hypomethylation of histone arginine residues in association with hyperhomocysteinemia may warrant further study. Another group has provided evidence of a more direct role of histone arginine in ischaemic stroke, in which levels of ADMA and symmetric monomethylarginine (SDMA) are highly expressed. 52 ADMA, a well-known NOS inhibitor, may not only contribute to reduced cerebral blood flow, but also oxidative stress and excitotoxicity-mediated neuronal death, to exacerbate damage from ischaemic stroke. 52 As such, the levels of ADMA could be a biomarker of ischaemic stroke damage. ADMA and SDMA levels have also been associated with the expression of inflammatory mediators following ischaemic stroke, such as monocyte chemotactic protein-1 (MCP-1), MMP-9, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), IL-6, C-reactive protein and S100B. 127 Interestingly, the expression of these mediators correlates with histone arginine methylation, 127 which could explain a relationship with inflammation in stroke.
Thus, while conventional therapeutics have been targeted for direct intervention against neutrophil transmigration without success, future studies might instead be redirected towards the modulation of critical histone writers and erasers to target stroke inflammation.
MicroRNAs and inflammation in stroke
MicroRNAs have been implicated in various aspects of stroke pathophysiology, including excitotoxicity, oxidative stress, apoptosis and inflammation. 60 The roles of miRNAs in regulating inflammation during stroke will be discussed here. In stroke patients with intracerebral haemorrhage, both spatial localization and expression levels of a wide array of miRNAs have been reported to be altered. 128 In adult rats subjected to focal cerebral ischaemia, expression of miR-124a, one of the more highly enriched miRNAs in the brain, was reduced in neural progenitor cells in the subventricular zone of the brain. 129 This expression pattern of miR-124a was accompanied by increased activation of Notch signalling. 127 Using an in vitro neural progenitor cell model, miR-124a was found to regulate stroke-induced neurogenesis with an involvement of Notch signalling. 129 Another study using a rat model of ischaemic stroke reported that miR-124 is upregulated in the plasma and may thus be a useful biomarker for stroke injury. 130 In human ischaemic stroke patients, blood levels of miR-30a and miR-126 were reduced for 24 weeks. 131 Interestingly, another microRNA, let-7b, was found to display a differential pattern of expression across different stroke subtypes. Let-7b was lower in stroke patients of a large atherosclerosis subtype compared to non-stroke patients, whereas in other types of ischaemic stroke, let-7b was higher than in healthy individuals. 131 The screening of different types of microRNAs may thus serve as a useful tool to assess the risks and prognosis of specific stroke subtypes. Indeed, expression of different microRNAs following stroke varies considerably in a tissue-dependent and stroke-subtype manner, coinciding with earlier findings that miRNAs work in a context-dependent setting. As such, future studies need to carefully consider the potential roles that these miRNAs may contribute to stroke before further targeted interventions are developed.
miRNAs have been demonstrated to regulate various aspects of thrombus formation, 132 which may thus contribute to the early phase of neuroinflammation following stroke. miR-19a, which has been reported to be reduced following ischaemic stroke, is associated with tissue factor pathway inhibitor (TFPI) and plasminogen activator inhibitor 1 (SERPIN1), as well as tissue factor III that modulates thrombus formation. 132 Furthermore, other prominent downregulated miRNAs that were associated with the modulation of other clotting mediators and identified to be altered in ischaemic stroke include miR-let-7i, miR-122 and miR-148. 132 Overall, downregulation of these miRNAs would be expected to promote blood clotting, and thus facilitate thrombus formation in the early phase of the inflammatory cascade during stroke. As such, therapies targeted at increasing levels of these miRNAs could prevent re-occlusion due to blood clotting.
Initiation of the inflammatory cascade occurs with the release of inflammatory mediators by resident brain cells such as microglia. In the ischaemic territory, these activated cells release mediators that propagate inflammatory damage. 60 Although these inflammatory mediators display potential for use as biomarkers of neuroinflammation and mediators of injury during stroke, therapeutics to target them have not been successful. However, recent studies have shown that miRNAs play an important role in modulating the expression and release of inflammatory mediators in a cell-dependent manner, which was previously poorly understood. miR-155 is well known to be a proinflammatory miRNA, and it is heavily involved in the inflammatory process during stroke. 133 It has been implicated as a regulator of macrophage differentiation and phenotype determination and its presence is necessary for macrophages to adopt a proinflammatory status. 133 Moreover, miR-155 is necessary to induce proinflammatory signalling in microglia, 134 where it is reported to downregulate suppressor of cytokine signalling 1 (SOCS-1) protein, resulting in microglial inflammation via upregulation of inflammatory molecules such as iNOS. 134 Inhibition of miR-155 using oligonucleotides reduces both inflammation by microglia and neuronal apoptosis. 134 Furthermore, miR-155 promotes secretion of both TNF and IL-1β via signalling of NF-κB and toll-like receptor 4 (TLR-4), and downregulation of myeloid differentiation primary response gene (MyD88). 135 Again, in mice following ischaemic stroke, treatment with the miR-155 antagomir, acetylbritannilactone, resulted in a reduced infarct volume through decreased expression of proinflammatory molecules, and a concomitant improvement in neurological score, thereby exerting a neuroprotective function. 136 As such, miR-155 is considered to be damaging during stroke, and future studies should seek to develop an antagomir that is translatable into humans. Another microRNA that is associated with proinflammatory effects in stroke is miR-210. 137 Treatment with an miR-210 inhibitor in mice subjected to MCAO stroke resulted in an overall decrease in expression of proinflammatory cytokines such as TNF, IL-1β and IL-6, as well as chemokines like CCL2 and CCL3. This was associated with reductions in cerebral infarct volume and neurological impairment. 137
Interestingly, it has been demonstrated that inflammasome activity may be regulated via the action of miRNAs. 138 The inflammasome is a cytosolic complex known to play a role in ischaemic stroke by promoting inflammatory and cell death mechanisms. 10 Roles for NLRP1 and NLRP3 inflammasomes and caspase-1 activation are well established in ischaemic stroke-induced neuroinflammation. 16 It was shown that following intracerebral haemorrhage, miR-223 can suppress the NLRP3 inflammasome by binding to its 3′ UTR sites. 138 As a result, both caspase-1 activation and IL-1β release were inhibited, leading to reduced brain oedema and improved neurological scoring following stroke. 138 That study provides novel insight in that microRNAs may not only regulate cytokine secretion directly, but also indirectly regulate their expression, such as through inflammasomes. As such, certain microRNAs seem to be closely associated with inflammatory responses in stroke and may reveal the mechanism behind the spatial and temporal control of these proinflammatory cytokines and chemokines action. Conversely, as compared to miR-155, miR-let-7a has been implicated in anti-inflammatory responses mediated by microglia during stroke. 139 miR-let-7a has been found to induce upregulation of anti-inflammatory mediators and recovery molecules, such as IL-4, IL-10, BDNF in microglia, as well as downregulate expression of proinflammatory iNOS and IL-6. 139 While miR-155 may regulate microglial inflammatory activity, other miRNAs, such as miR-let-7a, may regulate the opposite activity. However, notably, it has also been reported that miR-155 can increase expression of the neuroprotective cytokine, IL-10. 140 Nonetheless, it seems that miR-155 predominantly exerts proinflammatory effects in the brain during stroke. As such, microRNA agomirs and antagomirs that can modulate the levels of critical miRNAs are likely to be important for maintaining anti-inflammatory influences in the ischaemic brain territory. Indeed, many studies have attempted to inhibit critical miRNAs involved in maintaining neuroinflammation in stroke, with results being promising so far.137,141,142
It has been reported that microRNAs regulate the release of cytokines and chemokines, which activate endothelial cells and promote the extravasation and migration of neutrophils following cerebral ischaemia. 143 Indeed, certain microRNAs appear to be responsible for regulating endothelial cell activation and expression of adhesion molecules, to facilitate leukocyte adhesion. 143 For example, miR-146 is critical for controlling vascular inflammation by preventing endothelial activation via suppression of the NF-κB, mitogen activated protein kinase (MAPK) pathway, as well as downstream EGR transcriptional activity. 143 Moreover, miR-31 and miR-17-3P are each induced by TNF and antagonize expression of E-selectin and ICAM-1. 144 Inhibition of these microRNAs increased neutrophil adhesion to endothelial cells, whereas the microRNA mimetics prevented leukocyte adhesion. 144 miR-126 is a widely studied microRNA involved in vascular inflammation and angiogenesis, and which is a proposed biomarker for ischaemic stroke. 145 miR-126 is expressed by endothelial cells and inhibits VCAM-1 expression, thus modulating the level of vascular inflammation. 145 Furthermore, in a Chinese population the presence of a polymorphism at a single site affecting the miR-491-5p binding site may lead to an increased risk of cerebral ischaemia. 146 miR-491-5p antagonizes the expression of MMP-9, and so this alteration in binding may affect miR-491-5p activity and upregulate MMP-9 expression to facilitate blood–brain barrier disruption and leukocyte extravasation. 146 Another reported study indicated that a functional polymorphism in the 3′ UTR site of angiopoietin-1 gene leads to an alteration in the binding sites of miR-211, which in turn helps to reduce the risk of stroke occurrence. 147 In addition, miR-107 has been reported to target Dicer-1, which upregulates expression of vascular endothelial growth factor (VEGF), which should then facilitate post-stroke angiogenesis and promote recovery. 148 Thus, overall, different miRNAs appear to be able to differentially modulate various aspects of the maintenance of vascular function.
Epigenetic intervention in stroke
With the rise in numbers of studies focusing on epigenetic mechanisms, a revolution of information has emerged to inform a better understanding of neuroinflammation and neurotoxicity underlying stroke pathophysiology. Unfortunately, despite the elucidation of numerous molecular and cellular mechanisms that govern tissue injury following stroke, approved treatments for clinical stroke are limited to tissue plasminogen activator (tPA) and the recent introduction of mechanical thrombectomy. 149 However, the significant time limitations that apply to the use of these treatment strategies (i.e. <4.5 h for tPA and <8 h for thrombectomy in patients on blood thinners or who have received rTPA), means that >80% of ischaemic stroke patients are essentially left untreated, providing an enormous impetus for new stroke therapies to be developed.
Epigenetics represent a novel area in the study of stroke, and we have discussed that various epigenetic tags can be useful for predicting stroke risk, outcome and recovery. The epigenome varies considerably across individuals, and is heavily dependent upon the myriad of environmental stimuli that an individual is exposed to throughout their lifetime. Due to the diverse combinations of interplaying factors, every individual will have a unique epigenetic code, which may result in varying degrees of stroke risk, outcome and recovery. This concept may provide a better explanation for the interindividual variability that manifests across different stroke patients. A related aspect influenced by epigenome status may be the degree of inflammation occurring in individual stroke patients. If so, it is plausible that future therapies could be developed to modify the epigenomic status occurring during stroke to push it towards a state of regeneration rather than inflammation. Indeed, certain lifestyle factors have been shown to influence stroke risk, and as such there are likely to be good life practices associated with good health and the avoidance of stroke due specifically to epigenetic mechanisms. Such practices may involve modulation of an individual’s epigenetic tags to promote neuroprotection and tissue regeneration. In this last part of the review, we shall briefly highlight some recent findings with epigenetic drugs developed in animal models, and some lifestyle practices with potential to protect against stroke incidence and/or improve recovery.
Drugs to modulate epigenetic mechanisms in stroke
Given that DNA methylation profiles change considerably during stroke, a focus has been directed towards investigating the roles of DNMTs in stroke pathology. DNMTs have been found to be heavily involved in mediating changes in global DNA methylation status, and there have been studies assessing DNMT inhibitors for amelioration of stroke outcome. 76 For example, treatment of mice with a DNMT inhibitor, 5-aza-2′-deoxycytidine (also known as decitabine), conferred protection in a model of stroke. 76 However, this DNMT inhibitor appears to be effective only in the context of mild ischaemic damage, and the beneficial effect was not replicated in a stroke model of excitotoxic/necrotic cell death. 76 In a study using another DNMT inhibitor, zebularine, in a rat model of ischaemic stroke, this agent was reported to reduce neurological damage. 39 Both decitabine and zebularine are cytosine analogues that are nonspecific in their pharmacological actions, 39 which probably limits their usefulness in different stroke models and will slow their translational progress. As a result, there is a need to develop alternative drugs that are more specific, such as the MG98 antisense oligonucleotide, which shows specificity towards inhibiting mRNA translation of DNMT1.150–153 In addition, second-generation DNMT inhibitors, such as procainamide, hydralazine and VA, have also shown potential to modify the epigenetic machinery, which could thus be useful as stroke therapeutics.150–153 However, while targeting epigenetic regulation through DNA methylation may yield more promising results than conventional therapies, such drugs have still only been shown to inhibit DNMT. Notably too, a reduced level of DNMT1 in post-mitotic neurons may confer neuroprotection during cerebral ischaemia, and in the absence of DNMT1 using mutant studies, neuroprotection is abrogated in mice. 154 Thus, future research directed towards the development of DNMT inhibitors for stroke should consider the possible implications of total elimination of DNA methylation during stroke, including the broader complexities of DNA methylation epigenetics.
Besides DNMT inhibitors, it has been demonstrated more extensively that HDAC inhibitors can modify epigenetic programming in stroke and that this may ameliorate stroke injury.100,150,155–158 As discussed, prominent HDAC inhibitors in stroke research include VA, trichostatin A and sodium butyrate. These therapeutics have been widely tested in stroke models and have been consistently found to reduce ischaemic injury and to improve functional recovery.100,150,155–158 Besides the finding that HDAC inhibitors can modulate the histone acetylation machinery, it has also been discovered that HDAC inhibitors can attenuate proinflammatory pathways such as NF-κB and MMP-9 activity during stroke.100,150,155–158 The amelioration of the inflammatory response in stroke through reprogramming histone acetylation reveals great potential for the use of epigenetic drugs in the treatment of stroke. In conjunction with these more prominent HDAC inhibitors, other drugs such as SAHA (or vorinostat), sodium 4-phenylbutyrate and entinostat have also been reported to provide neuroprotection against experimental stroke injury.105,159,160 Modulation of histone acetylation status might therefore be able to alleviate inflammation during stroke, as well as modulate a myriad of other relevant protective mechanisms.
With a sufficient level of understanding of the specificity of various miRNAs and their substrates, there is optimism about the plausibility of developing miRNA-epigenetic drugs. Indeed, the development of synthetic miRNA mimetics has already been employed in many studies, and has illuminated the spatial and temporal regulation of inflammation during stroke. As mentioned, miRNA agomirs are synthetic mimetics that perform a similar function to that of biological miRNAs in vivo, whereas miRNA antagomirs act as inhibitors that prevent the action exerted by specific miRNAs.161,162 miRNAs that are neuroprotective tend to be downregulated in stroke, whereas other miRNAs that drive toxic inflammatory responses tend to be upregulated (Table 1). As such, future studies should assess the effects of delivering miRNA agomirs and/or antagomirs, as appropriate, to promote a neuroprotective microenvironment during stroke. In such a manner, miRNAs could serve as a regenerative medicine to be delivered to stroke patients, although until now there has been limited development in animals due to low delivery efficiency, bioavailability and half-life, as well as some cytotoxicity,163–168 and no studies have yet been performed in humans. It is too early to speculate as to the likely success of such an approach, but future studies might consider the general concept of targeting miRNA biosynthesis and degradation in vivo system to modulate the levels of key miRNAs.
Epigenetic Modifications Involved in Stroke.

Conclusion
Stroke continues to be a major cause of death globally, and despite extensive research into the molecular and cellular mechanisms of its pathogenesis, the use of tPA administration and/or mechanical thrombectomy involve strict eligibility criteria that greatly limit their usefulness. While many molecular targets have been identified to mediate inflammation during stroke, very few clinical trials have provided evidence to support positive effects of anti-inflammatory treatment strategies in acute ischaemic stroke.169,170 The lack of major progress in these research attempts could be in part due to a lack of understanding of the spatial and temporal regulation of key molecular players in the inflammatory cascade. Further, owing to interindividual variation in the degree of vulnerability and extent of damage induced during stroke, it has been suggested that the epigenome status of an individual may be relevant for their ensuing stroke pathology. Indeed, through DNA methylation, histone modifications, and miRNAs, inflammatory mediators can be regulated in an epigenetic context. Epigenetic regulation also provides a rational explanation as to why stroke is such a complex disease, with the possibility that overall epigenetic tags are created from the interplay of multiple genes and environmental factors. This may partly explain why findings from animal models may not be translatable to human subjects as their epigenomes will differ greatly. As such, this review proposes the exploration of an epigenetic approach to intervention for stroke, involving not only epigenetic-related drugs, but also positive lifestyle practices such as dietary restriction and healthy eating.
Footnotes
Funding
This work was supported by the National University of Singapore strategic grant (Memory Network Program: R-185-000-271-646), National Medical Research Council Research Grants (NMRC-CBRG-0102/2016), Singapore National Medical Research Council Research Grants (NMRC/OFIRG/0036/2017) and the National Health and Medical Research Council of Australia (APP1079467; APP1064686; APP1085323).
Conflict of interest statement
The authors declare that there is no conflict of interest.