
Abstract
Background:
Both the United States (US) Food and Drug Administration (FDA) and the European Union (EU) European Medicines Agency (EMA) order pediatric clinical trials as a condition for approval of new compounds. We evaluate clinical value and likelihood of sufficient recruitment for pediatric multiple sclerosis (pMS) studies and discuss US and EU pediatric legislation with pMS as a paradigm.
Methods:
We analyzed pMS clinical trials requested by the FDA and the EMA and industry-sponsored pMS studies registered on www.clinicaltrials.gov and www.clinicaltrialsregister.eu.
Results:
The FDA demands four and the EMA 15 pMS trials
Conclusions:
pMS is rare. Neither FDA nor EMA prioritize compounds for potential benefit in pMS. The EMA in particular orders multiple pMS studies, which will probably not recruit enough patients. Therefore, it is likely that the pMS trial outcomes will not be relevant for evidence-based medicine analyses, clinical practice and a pMS label for the respective drug. EMA requests for multiple pediatric studies have been described in metastasized adolescent melanoma, another very rare pediatric disease. The terms ‘ghost studies’ and ‘therapeutic hostages’ have been proposed for such trials and children whose parents are lured into permitting study participation. Clinical studies are not ethical if the probability is high that they will not provide reasonable outcomes. For now, pMS clinicians will have to continue to use new MS drugs in children off-label. They might consider a more proactive international coordinating role in prioritizing and testing new MS compounds in children.
Keywords
Introduction
Pediatric multiple sclerosis (pMS) occurs in a small fraction of patients with multiple sclerosis. About 2–5% of patients with MS are under 18 years old, less than 1% are under 10 years old, and the incidence is less than one in 100,000 people [Chou et al. 2014; Pena and Lotze, 2013; Suppiej and Cainelli, 2014; Waldman et al. 2014]. No therapies are approved for pMS by the US Food and Drug Administration (FDA), and only limited interferon use by the European Medicines Agency (EMA) [Chitnis et al. 2013]. Modern drug labels result from pharmaceutical legislation in 1962 [Rose, 2012]. This also initiated a discussion on ‘off-label’ use in children and children’s status as ‘therapeutic orphans’ [Rose, 2008, 2014a, 2014b; Shirkey, 1968]. Since 1962, pharmaceutical companies must prove safety and efficacy (S&E) for drug approval in adequate clinical trials, while before, they could make claims for their products without scientific proof. After approval, physicians can use drugs off label in other disorders or patient populations, while pharmaceutical companies must not promote off-label use. However, they may sponsor further trials, and if the outcomes are positive, request expanded approval. This works well in frequent disorders, but is challenging in rare diseases or small patient cohorts like pMS. Newer immunotherapies offer advantages in S&E in adult MS [Coles, 2015]. The International Pediatric Multiple Sclerosis Study Group (IPMSSG) has addressed essential challenges of pediatric clinical studies in new MS drugs in the wake of US and European Union (EU) pediatric legislation [Chitnis et al. 2013], both introduced to change children’s status as ‘therapeutic orphans’. We investigated to what degree the US- and EU-mandated pMS clinical studies are practicable and have clinical value. This is of relevance beyond the US and EU, as pediatric clinical trials recruit patients on a global level today [Pasquali et al. 2010].
Methods
All FDA-approved MS drugs were checked for pediatric study requests in the corresponding FDA approval letters (available on the internet). All EMA requests for pediatric clinical trials for MS drugs within the respective pediatric investigational plan (PIP) were evaluated. PIPs are published in a shortened version on the EMA website. The titles of all mandated clinical studies and other measures are listed towards the end of the published PIP. In addition, we analyzed existing clinical MS trials, registered on ClinicalTrials.gov and ClinicalTrialsRegister.eu.
As all information used for this paper is published or directly available on the internet, no ethics board approval was needed for this paper.
Results
Table 1 and Figure 1 describe key milestones in the development of modern pharmaceutical legislation, consequences for its use in children, including the question of off-label use, and the background of US and EU pediatric legislation. Table 2 provides a schematic overview of US and EU pediatric legislation.
Key dimensions of use of drugs in children.


Timelines of key dimensions in use of modern drugs in children.
US and EU pediatric legislation.

CHMP, Committee on Human Medicinal Products; FDAMA, US Food and Drug Administration Modernization Act; FDASIA, US Food and Drug Administration Safety and Innovation Act; PDCO, pediatric committee; PREA, Pediatric Research Equity Act; PIP, European Union pediatric investigation plan; PSP, US Food and Drug Administration pediatric study plan.
Table 3 lists the studies demanded by the FDA in children and adolescents with MS. S&E trials were mandatory for four compounds. The other MS compounds were introduced when the FDA could not yet demand pediatric trials. The FDA ordered additional pharmacokinetic (PK) data for some compounds for children and in all trials comparison of the respective medication against an appropriate control. The deadline of the last study report of the FDA-demanded pMS trials is in 2021.
FDA-required pediatric MS studies.

AC, active controlled; C, controlled; E, efficacy; FSR, final study report; IFβ, interferon β; MD, multiple dose; MS, multiple sclerosis; PG, parallel group; PK, pharmacokinetics; R, randomized; S&E, safety and efficacy.
Table 4 shows the PIP-mandated pMS studies. PIP studies for natalizumab and alemtuzumab were waived. One to two comparisons to active control therapies are requested for all other new compounds, and for some compounds, PK data. Most trials have to compare compounds either against an appropriate comparator or interferon β. A placebo-controlled trial is demanded for teriflunomide (Table 3, study 13). The deadline of the last EMA/pediatric committee (PDCO)-demanded clinical trials is in 2024. Negotiations with the authorities to establish a new PIP lasts for approximately 1 year; for a PIP modification, approximately 6 months [Rose, 2012, 2014b].
Multiple sclerosis PIP-requested clinical studies.
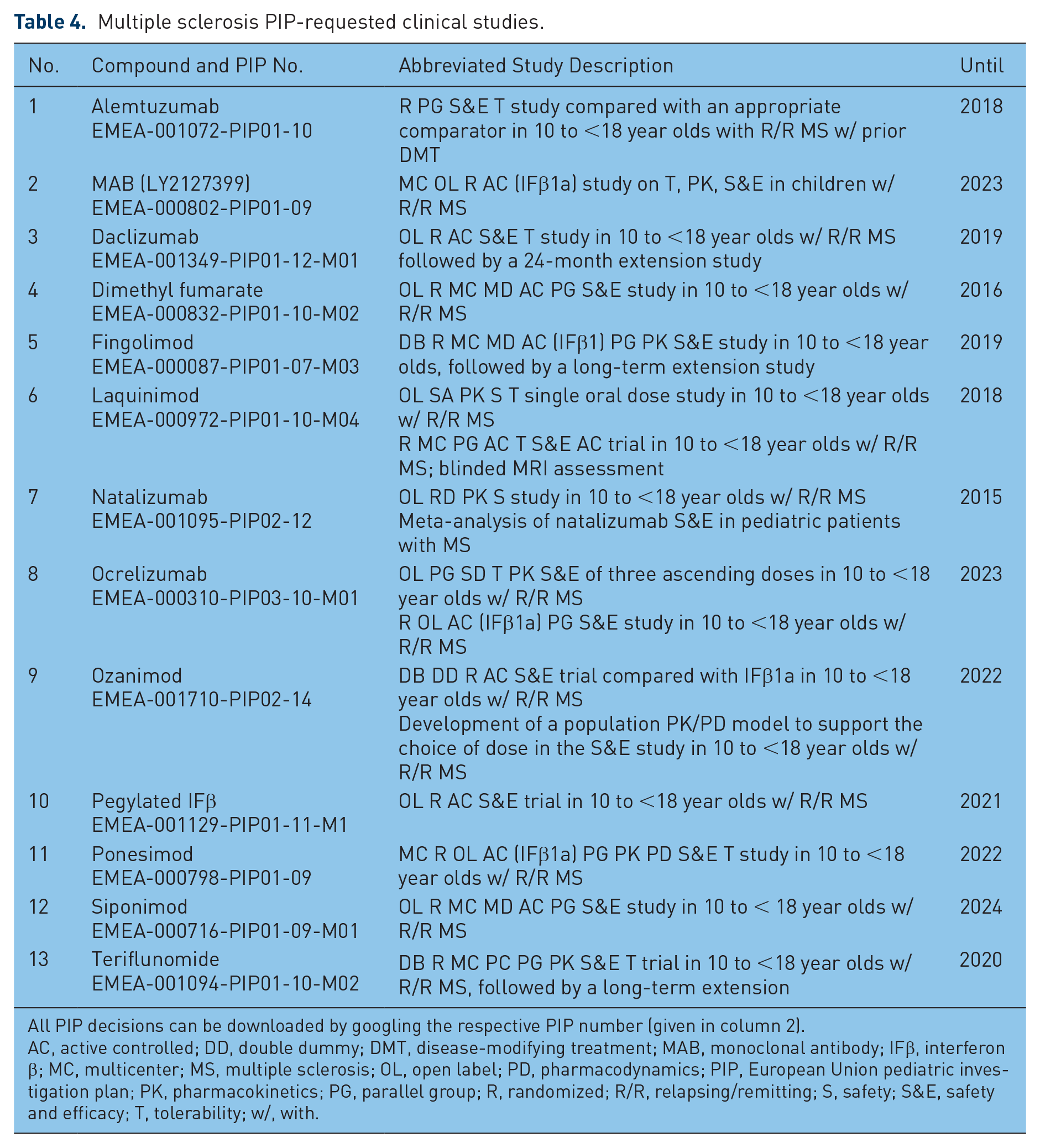
All PIP decisions can be downloaded by googling the respective PIP number (given in column 2).
AC, active controlled; DD, double dummy; DMT, disease-modifying treatment; MAB, monoclonal antibody; IFβ, interferon β; MC, multicenter; MS, multiple sclerosis; OL, open label; PD, pharmacodynamics; PIP, European Union pediatric investigation plan; PK, pharmacokinetics; PG, parallel group; R, randomized; R/R, relapsing/remitting; S, safety; S&E, safety and efficacy; T, tolerability; w/, with.
Table 5 shows the registered industry-sponsored pMS trials on ClinicalTrials.gov. The placebo-controlled trial with teriflunomide (Table 5, number 3) is probably PIP triggered. Studies 2 and 6 in Table 5 might be triggered by both the FDA and EMA. Study 4 in Table 5 compares dimethyl fumarate with placebo, whereas both the FDA and EMA/PDCO ask for an active control. The databases ClinicalTrialsRegistry.eu and ClinicalTrials.gov show four identical trials. No further pMS trials were identified on ClinicalTrialsRegistry.eu.
Industry-sponsored pediatric MS studies on ClinicalTrials.gov.

The search terms ‘multiple sclerosis children’ in ClinicalTrials.gov gave 186 studies on 18 January 2016, of which 11 were industry-sponsored trials in children. Trials without patients’ age were not entered into this table.
The search terms ‘multiple sclerosis AND children’ in ClinicalTrialsRegister.eu (EudraCT) gave 11 results, of which 4 studies corresponded to trials on ClinicalTrials.gov. The other trials were academic trials or trials with adults only
Every single study listed in this table can be looked at directly by googling the NCT number.
DM, fumarate dimethyl fumarate (BG00012); IFβ1a, interferon β1a; IRBP, immune response BioPharma; MRI, magnetic resonance imaging; MS, multiple sclerosis; NR, not reported; PhI, phase I; PK, pharmacokinetics; S&E, safety and efficacy.
Discussion
We showed regulatory complexity and bureaucratic effort for MS compounds that are already approved (FDA) or for which an approval is planned (EMA).
The situation is peculiar in the case of natalizumab. Its use is ‘contraindicated in patients below the age of 18 years’ [Kornek, 2015]; however, only in the EU. In other words, in the EU children with MS have to wait until their 18th birthday before they may receive natalizumab, despite reports on S&E in minors [Kornek, 2013]. Here the EMA perceives everything as forbidden that has not been granted approval. Thus the EMA neglects individual calculations of risk/benefit ratios, the responsibilities of the treating physician and the increasingly well informed patients and parents. There is also a discrepancy of attitudes by the FDA and EMA. The FDA judges the use of natalizumab in children with MS as ‘not indicated’ [FDA, 2012] in contrast to the contraindication by the EMA. In general, both the FDA and the EMA list contraindications as conditions that may represent a lethal threat.
The IPMSSG stated in 2013 that the number of existing pMS cases worldwide would allow concurrent performance of one to two trials only [Chitnis et al. 2013]. Today, we have four FDA- (Table 3) and 15 EMA-demanded pMS clinical trials (Table 4). As a result, recruitment for all these studies is difficult or perhaps may never be completed.
The EMA’s predominant focus on formal regulatory aspects is reflected in numerous documents published by the EU commission, the EMA and members of the EMA PDCO. Key features of this thinking are listed in Table 6.
Theoretical foundations of the European Union pediatric legislation.

EMA, European Medicines Agency; EU, European Union; PDCO, pediatric committee.
One conclusion of the IPMSSG summit [Chitnis et al., 2013, p. 1165] was that theoretically ‘placebo-controlled trials (PCTs) are ideal in the current environment as there are no approved treatments for pediatric MS, and placebo-controlled trials typically allow for smaller sample sizes than superiority trials’. In practice, there will be problems in getting a positive ethical vote and recruiting pMS cases for studies that include placebo treatment. There is enough evidence that, for example, interferons and glatiramer acetate are superior to placebo in adult patients with MS [Coles, 2015; Pena and Lotze, 2013; Kornek, 2015]. It would be irresponsible and unethical of the treating physician not to offer ‘off-label’ use with the consent of the patient and the parents. The 18th birthday is a legal limit, but not a biological or medical one. Clinical trials in children and adolescents are necessary, but to demand S&E trials in pMS for every single new compound is questionable, as pMS is too rare. Here the academic clinical community should scrutinize the position of regulatory authorities. The American Academy of Pediatrics has published a clear positive statement on the legitimacy of off-label use in children in general [Fratarelli et al., 2014]. The IPMSSG agreed that ‘a stepwise approach to the launch of clinical trials for the most promising medications is necessary in order to ensure study completion’ [Chitnis et al 2013]. The regulatory authorities do not prioritize the incoming flow of new effective medicines but instead enforce many clinical trials in pMS. Specifically the EMA-mandated trials might become ‘ghost studies’: trials are initiated, study sites opened, protocols submitted to international review boards, but recruitment fails [Rose, 2014c; Rose and Senn, 2014; Rose and Kummer, 2015]. Within 5–10 years, pharmaceutical companies will request a PIP modification from the EMA. An evaluation will then be limited to the few patients whose parents have been lured to allow their children to participate. These study participants may be looked upon as ‘therapeutic hostages’ [Rose, 2014c; Rose and Senn, 2014; Rose and Kummer, 2015].
Conclusion
We have shown that specifically the EMA forces pharmaceutical companies to initiate questionable clinical trials in pMS. Instead, prioritization is needed. This is a new challenge in our increasingly globalized world. Advocacy groups, pMS specialists and parents of children with MS should consider an international initiative to establish a framework for realistic and reasonable pediatric investigations in new powerful MS drugs. It would be desirable for the IPMSSG to play a key role.
Footnotes
Acknowledgements
Klaus Rose: study concept, acquisition of data, first analysis, first draft manuscript. Thomas Müller: study concept, review of data analysis, critical revision of manuscript.
Authors’ note
Klaus Rose has worked for 20 years in pharmaceutical industry in drug development. Independent since 2011, he consults on pediatric drug development, organizes scientific conferences, edits books, and publishes. His clients are small, medium-size and large pharmaceutical companies. He is also father of a daughter with a rare disease. Thomas Müller is chief medical officer. He has been a clinician all his professional life.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors have no conflicts of interest to declare.