
Abstract
Background
Treatment options for children and adolescents with migraine are limited. This study evaluated rizatriptan for the acute treatment of migraine in children and adolescents.
Methods
Randomized, double-blind, placebo-controlled, parallel-group trial in migraineurs 6–17 years old with unsatisfactory response to nonsteroidal anti-inflammatory drugs or acetaminophen/paracetamol. The trial included a double-blind run-in with weight-based rizatriptan dosing (5 mg for <40 kg, 10 mg for ≥40 kg). In the Stage 1 run-in, patients were randomized in a ratio of 20:1 placebo:rizatriptan and were instructed to treat within 30 minutes of a moderate/severe migraine. Patients with mild/no pain after 15 minutes of treatment (responders) took no further study medication, whereas patients with moderate/severe pain (non-responders) proceeded to take study medication in Stage 2. Non-responders who received placebo in Stage 1 were randomized 1:1 to rizatriptan:placebo, whereas non-responders who received rizatriptan in Stage 1 were allocated to placebo in Stage 2. The primary efficacy endpoint was pain freedom at 2 hours after Stage 2 dose in 12–17-year-olds.
Results
A higher proportion of 12–17-year-olds on rizatriptan had pain freedom at 2 hours compared with those on placebo: 87/284 (30.6%) versus 63/286 (22.0%), odds ratio = 1.55 [95% CI: 1.06 to 2.26], p = 0.025. Adverse events within 14 days of dose in 12–17-year-olds were similar for rizatriptan and placebo. The pattern of findings was similar in 6–17-year-olds.
Conclusion
Rizatriptan demonstrated a statistically significant improvement over placebo in eliminating pain and was generally well tolerated in migraineurs aged 12–17 and 6–17 years.
Trial Registration
ClinicalTrials.gov NCT01001234
Introduction
Migraine affects approximately 8% of children and adolescents (1,2). For some, acute treatment with standard analgesics such as acetaminophen (referred to as paracetamol in the EU) and ibuprofen can provide adequate relief (3,4), but many young migraineurs do not gain sufficient relief with these treatments. Compared with adults, the number of drugs studied and approved for the abortive treatment of migraine attacks in children and adolescents is limited (5). This is due both to the practical challenges of conducting studies in children and the difficulty of demonstrating efficacy of treatments in young migraineurs (6). Nonetheless, sumatriptan nasal spray in the EU (7) and almotriptan tablets in the US (8) are approved for the acute treatment of migraine in 12–17-year-olds. Recently, rizatriptan was approved by the US Food and Drug Administration for the acute treatment of migraine in 6–17-year-olds on the basis of the study described in this paper.
Studies of acute migraine therapies have failed for a number of potential reasons. There is typically a greater response to placebo in children than adults (9,10) and the short duration of migraine attacks in some children may contribute to this observation. It is also possible that children may be more susceptible to suggestion, and therefore respond to what they perceive as adult expectations about response to medication. Children typically receive a lower dose than adults, raising the possibility of underdosing. Lack of developmentally appropriate procedures (for example not using age-appropriate assessment instruments) and use of adult diagnostic criteria for children may be other explanations for study failures.
A number of previous randomized controlled studies have evaluated rizatriptan 5 mg in adolescent migraineurs (the standard adult dose is 10 mg) and found that it was not more effective than placebo at eliminating or reducing pain as assessed using the standard 4-grade verbal pain scale from adult studies (11,12). Most recently, Ahonen et al. (13) performed a three-way crossover randomized controlled study in 6–17-year-olds using a weight-based rizatriptan dosing regimen of 5 mg for those <40 kg and 10 mg for those ≥40 kg. The primary efficacy endpoint of 2-hour pain relief, defined by a decrease of two grades on a 5-Face Pain Scale (14), was reached in significantly more patients in the rizatriptan group than the placebo group. The dosing regimen was generally well tolerated. This weight-based dosing regime is supported by data from a pharmacokinetic study in pediatric migraineurs (15) that demonstrated AUC and Cmax values similar to those historically observed in adults administered rizatriptan 10 mg (16). These findings suggest that weight-based rizatriptan dosing may provide an optimized regimen in pediatric migraineurs.
The aim of the present study was to further evaluate the acute treatment of migraine with rizatriptan versus placebo in children and adolescents who had not obtained adequate response to nonsteroidal anti-inflammatory drugs (NSAIDs) and acetaminophen using a weight-based dosing schedule and a double-blind placebo run-in design.
Methods
Patients
Participants were males and females aged 6–17 years who were ≥20 kg in weight and who had at least a 6-month history of migraine attacks with or without aura as defined by International Headache Society criteria (17), usually lasting 3 hours or more (when untreated). Patients had ≥1 and ≤8 moderate to severe migraine attacks with or without aura per month in the 2 months before the screening visit, and had not, by history, experienced satisfactory relief with NSAIDs or acetaminophen. Patients were excluded if they had not experienced satisfactory relief from migraine pain during prior treatment with two or more adequate courses of triptans.
Standard protocol approvals, registrations, and patient consents
The study was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies.
Each patient provided assent and their parent/caregiver provided written informed consent. The trial was registered at ClinicalTrials.gov (NCT01001234). A Scientific Advisory Committee comprising non-Merck and Merck scientists contributed to the development of the protocol, statistical analysis plan, analysis and interpretation of the data, and authoring of the manuscript. The Scientific Advisory Committee members are listed as authors of this paper.
Study design and procedures
This randomized, double-blind, placebo-controlled, parallel-group outpatient study (Merck Protocol 082) was performed at 191 sites (134 sites in the United States and 57 sites in Europe, India, and Canada) from December 2009 to April 2011. Enrolment was competitive with the proviso that enrolment at a single site was limited to approximately 10% of the total sample. Patients attended an initial clinic screening visit to assess eligibility and undergo routine physical examinations, vital signs, laboratory screens, and ECGs. Eligible patients subsequently treated a single moderate or severe migraine attack in two stages, with the purpose of Stage 1 being to identify placebo non-responders who would then enter into Stage 2. Rizatriptan dosing throughout the study was weight-based: patients <40 kg received 5 mg rizatriptan as an orally disintegrating tablet (ODT; referred to as oral lyophilisate in the EU) and those weighing ≥40 kg received 10 mg rizatriptan ODT. In Stage 1, patients were randomized in a 20:1 ratio to placebo ODT:rizatriptan ODT, with randomization stratified based on age (6–11 years versus 12–17 years; these age groupings were intended to roughly define pre-pubertal and pubertal populations). Enrolment for the 12–17 years group in Stage 1 included similar numbers of patients in the 12–14 and 15–17 age groups. Patients were instructed to administer study medication within 30 minutes of onset of a qualifying migraine attack (see below). Fifteen minutes after taking study medication, patients called into an interactive voice-response system to report their pain intensity level. Patients who reported mild or no pain (responders) were instructed to take no further study medication. Patients who reported moderate or severe pain (non-responders) were instructed to take study medication in Stage 2. Non-responders to placebo in Stage 1 were randomized 1:1 to rizatriptan:placebo, with randomization stratified by age (6–11 years versus 12–17 years) and migraine intensity reported at 15 minutes after Stage 1 dose (moderate versus severe). Non-responders to rizatriptan in Stage 1 were allocated to receive placebo in Stage 2. Patients who did not obtain satisfactory relief of migraine pain at 2 hours after Stage 2 treatment could treat with their usual headache therapy at that time, or any time thereafter, if the headache did not resolve or recurred, with the exception that triptans and ergot derivatives were prohibited for 24 hours following the last dose of study treatment. A follow-up clinic visit was performed within 14 days after dosing with study medication to review patient diaries and perform routine safety assessments.
A qualifying migraine was defined as a migraine of moderate or severe intensity that: was not spontaneously resolving; was not preceded by the use of ergot-type medications or triptans within the previous 24 hours or antiemetic and analgesic medications within the previous 6 hours; was not preceded by another migraine or other headache within the last 48 hours; and occurred in a setting in which study medication and procedures could be properly administered (that is, it was possible to administer study medication within 30 minutes of moderate/severe pain onset and the patient was capable of staying awake for at least 2 hours after administration of the Stage 2 dose to complete the migraine diary). If the patient was ≤12 years of age (or if deemed necessary by the patient’s parent/guardian for older children), a responsible adult who had knowledge of the conduct of the study (e.g., a school nurse) had to be available to supervise migraine treatment and study procedures.
Details of randomization, treatment allocation, and blinding are provided in the study protocol (see Supplemental online material). All study personnel, including investigators, study site personnel, patients, and Merck staff involved in the study, remained blinded to treatment allocation throughout the study. Unblinding took place after data collection was complete.
Assessments
A paper migraine diary was used to evaluate outcomes. Headache pain intensity on the 5-Face Pain Scale (14), presence of associated symptoms (nausea, vomiting, phonophobia, photophobia), and degree of functional disability on a 4-grade scale (able to complete daily activities ‘as usual’, ‘some’, ‘a little’, or ‘not at all’) were assessed at Stage 1 at 0 minutes (immediately before administration of Stage 1 study medication), at 15 minutes after Stage 1 dose (Stage 2 baseline - immediately before administration of Stage 2 study medication), and at 0.5, 1, 1.5, 2, 24 and 48 hours after Stage 2 dose. For the 5-Face Pain Scale (14), patients were instructed to place an ‘X’ on the face corresponding to their current level of pain. Faces were mapped to the following verbal descriptors: Face 5 = ‘severe pain’, Faces 4 and 3 = ‘moderate pain’, Face 2 = ‘mild pain’, Face 1 = ‘no pain’. Any side-effects experienced after taking study medication were recorded in the patient diary. Other migraine medication, taken up to 24 hours before administering study medication and up to 48 hours after administering study medication, was recorded in the diary. At 24 hours after Stage 2 dose, patients recorded their overall level of satisfaction with treatment on a 5-point scale (medication made the patient feel ‘a lot better’, ‘a little better’, ‘did not do anything’, ‘a little worse’, ‘a lot worse’). Parents/caregivers could assist the patient in the reading and comprehension of the diary, or by acting as the scribe, but were instructed not to complete the 5-Face Pain Scale for the patient.
Tolerability and safety were assessed via adverse event reports and physical and laboratory examinations, ECGs and vital signs performed before and after the study.
Statistical analysis
There was one primary hypothesis (pain freedom in 12–17-year-olds) and three secondary hypotheses (pain relief in 12–17-year-olds, pain freedom in 6–17-year-olds, and pain relief in 6–17-year-olds), all at 2 hours after Stage 2 dose. Pain freedom was defined as a reduction in pain from moderate or severe (Face 3, 4, or 5 on the 5-Face Pain Scale) to no pain (Face 1) and pain relief was defined as a reduction in pain from moderate or severe (Face 3, 4, or 5) to mild or no pain (Face 2 or 1). A post hoc efficacy analysis was conducted using the same endpoint as that used in the Ahonen et al. study (13) in which 2-hour pain relief was defined as a reduction of at least 2 grades from a baseline pain score of moderate or severe (Face 3, 4, or 5). Analysis of efficacy data was based on the Full-Analysis-Set (FAS) population, which included patients who did not respond to placebo at Stage 1 and were randomized to Stage 2. Additionally, the FAS population also required patients to have taken the Stage 2 study medication, have a moderate or severe Stage 2 baseline score, and have at least one post-Stage-2 dose efficacy measurement before or including the 2-hour time point. Efficacy for all primary and secondary endpoints was assessed using a logistic regression model adjusting for Stage 2 baseline migraine severity and geographical region. For the secondary hypotheses involving the combined 6–17 year age range, the age group (6–11 or 12–17-year-olds) was also included as a covariate. Exploratory efficacy endpoints included sustained pain freedom from 2 to 24 hours (defined as pain freedom at 2 hours with no recurrence of pain and no use of additional migraine medications from 2 to 24 hours), sustained pain freedom from 2 to 48 hours, absence of photophobia, phonophobia, nausea, and vomiting, ‘as usual’ rating of functional ability, use of rescue medication, and satisfaction with treatment at 24 hours (based on a dichotomization of the 5-point scale, with ‘satisfied’ corresponding ratings of ‘a lot better’ or ‘a little better’). Exploratory endpoints were evaluated in a similar manner to the primary and secondary endpoints. To control for multiplicity, testing from the primary hypothesis to the three secondary hypotheses was performed in a sequential manner. Further details are provided in the study protocol (see Supplemental online material). All other endpoints were considered exploratory and no multiplicity adjustment was made. Results for exploratory endpoints characterized as statistically significant are in reference only to nominal significance (unadjusted p-value < 0.05).
The All-Patients-as-Treated (APaT) population was used for the analysis of safety data. The APaT population consisted of all randomized patients who received at least one dose of study treatment (patients who only participated in Stage 1 were also included). Patients were included in the treatment group corresponding to the study treatment that they actually received, with active treatment taking precedence over placebo treatment. Thus, patients who took any rizatriptan during the study (Stage 1 or 2) were included in the active treatment group and patients who only took placebo during the study were included in the placebo group. The numbers of patients with any adverse events, drug-related adverse events, serious adverse events, and triptan-related adverse events (asthenia, dizziness, dry mouth, fatigue, chest discomfort, throat tightness, myalgia, nausea, paraesthesia, somnolence) as well as specific adverse events occurring in ≥4 patients in any treatment/age group, were tabulated by treatment group. Safety and tolerability were also assessed by review of laboratory values, ECGs, and vital signs.
Power
This was a single study, powered for the older age group (12–17-year-olds) and the combined younger and older age groups (6–17-year-olds). The study was not powered for the younger age group (6–11-year-olds), but a statement about the size of effect that could be detected at a nominal 0.05 alpha level with the planned sample size for 6–11-year-olds was pre-specified in the protocol.
The initial plan was to enroll approximately 900 12–17-year-olds in Stage 1 to yield approximately 548 evaluable adolescent patients in Stage 2. Based on this planned sample size the study had 80% power (2-sided, α = 0.05) to demonstrate superiority of rizatriptan versus placebo in the percentage of patients with 2-hour pain freedom, assuming a treatment difference of 11 percentage points (36% versus 25%) for the percentage of patients with 2-hour pain freedom. An interim efficacy analysis was planned for 12–17-year-olds to determine whether any modifications needed to be made to the sample size, using methods that maintained control of the Type I error. The results from the interim efficacy analysis, along with available safety data, were reviewed by an independent, external Data Monitoring Committee that was responsible for making enrolment recommendations based on this pre-planned interim analysis. Further details are provided in the study protocol (see Supplemental online material).
For 6–11-year-olds, the plan was to enroll approximately 340 patients in Stage 1 which was expected to yield approximately 160 evaluable patients in Stage 2. For a fixed sample size of 160 evaluable patients this study had 80% power (2-sided, α = 0.05) to demonstrate nominal superiority of rizatriptan over placebo in the percentage of patients with 2-hour pain freedom if the true underlying treatment difference was approximately 21 percentage points (41% versus 20%).
Results
Patient accounting and demographics
The trial profile is summarized in Figure 1 and full details of patient accounting and exposure to study medication are shown in Supplementary Tables 1 and 2. Of the 1382 randomized 6–17-year-olds, 977 took study medication. The main reason that patients did not treat was lack of a qualifying migraine attack (261/405, 64.4%). Of the 977 patients who treated with study medication, 894 (91.5%) completed the study. Among patients who treated with study medication, the primary reason for study discontinuation was a protocol violation (74/83, 89.2%) because they did not follow/complete the required study procedures. The number of enrolled patients per site varied from 1 to 140, with most sites (182/191) enrolling fewer than 20 patients and only one site enrolling more than 29 patients.
Study flowchart. This flow chart shows a high-level summary of the numbers of patients 6–17 years of age at key stages of the study and that were included in the efficacy and safety analyses. *There were 14 patients (seven for rizatriptan and seven for placebo) who treated only in Stage 2. **Patients could be excluded for more than one reason. †Patients were analyzed for safety according to the treatment they received; in some cases patients received a different treatment from that to which they were randomized. See Supplementary Tables 1 and 2 for full details of patient accounting and patient exposure to medication. Patient characteristics by treatment for the 6–17 year age group (all patients treated). NA: patient was not treated in Stage 2; that is, patient was a responder who had mild or no pain 15 minutes after dosing in Stage 1. Patient self-reported data on a migraine history questionnaire administered at baseline. Stage 2 baseline migraine characteristics for the 6–17 year age group (all patients treated with Stage 2 medication).* Data shown are characteristics of the migraine immediately before Stage 2 dosing, or 15 minutes after initial dosing in Stage 1. There was a small amount of missing data for most measures (maximum of 0.5%).

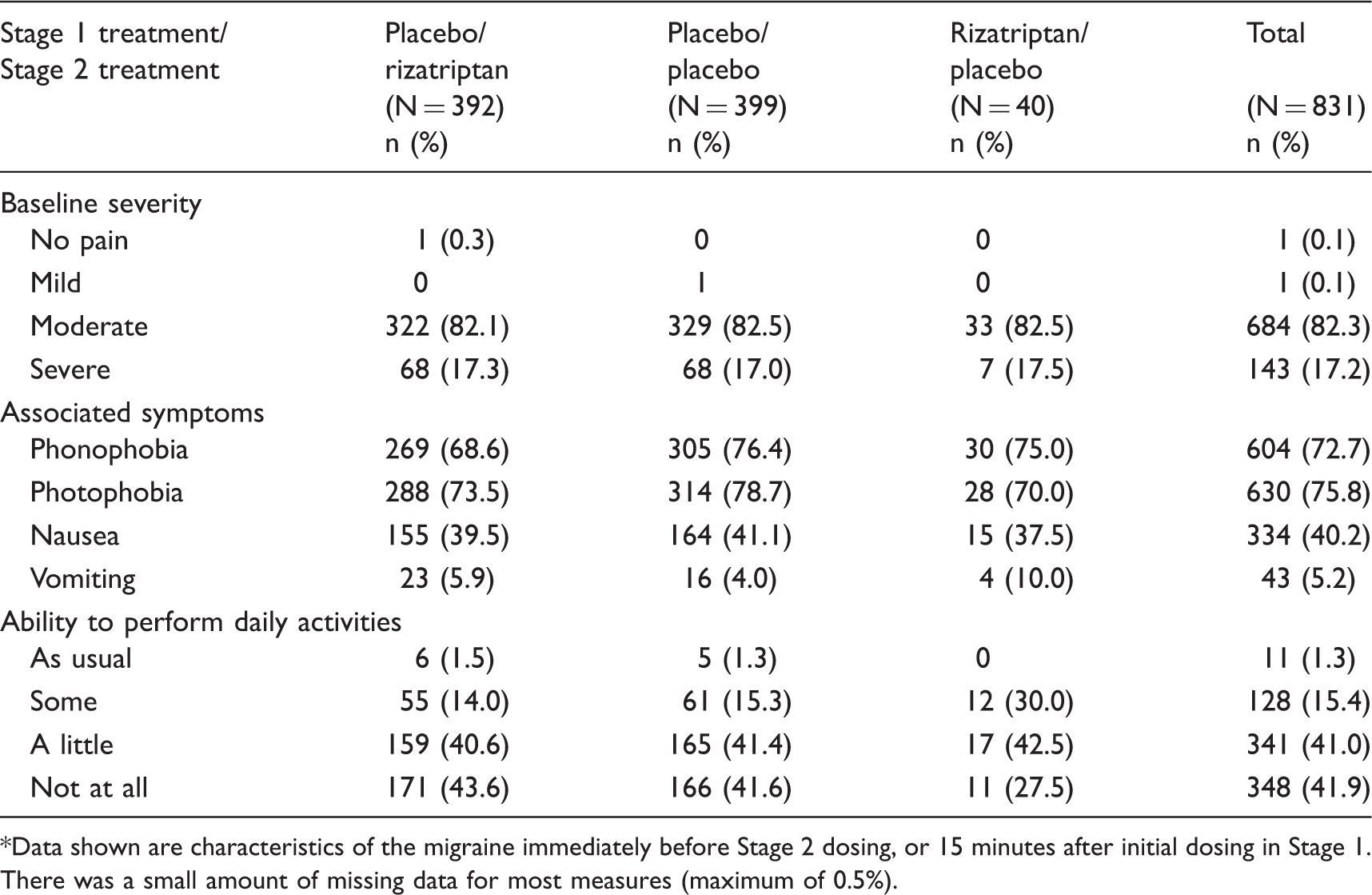
The characteristics of 6–17-year-olds who took study treatment are shown in Table 1. Of the 977 treated patients, 550 (56.3%) were females, 702 (71.9%) were 12–17-year-olds and 275 (28.1%) were 6–11-year-olds. The proportion of females was higher in the 12–17-year-olds (428/702, 61.0%) than in the 6–11-year-olds (122/275, 44.4%). Most 6–17-year-olds (731, 74.8%) weighed ≥40 kg; of the 702 12–17-year-olds 642 (91.5%) weighed ≥40 kg and 60 (8.5%) weighed <40 kg, while of the 275 6–11-year-olds 89 (32.4%) weighed ≥40 kg and 186 (67.6%) weighed <40 kg. Most of the 977 6–17-year-olds (718, 73.5%) were triptan-naïve and the most common usual migraine treatments were NSAIDs (586, 60.0%) and acetaminophen (435, 44.5%).
A total of 831/977 (85.1%) 6–17-year-olds treated in Stage 2. The characteristics of the treated migraine attacks for 6–17-year-olds who entered Stage 2 are shown in Table 2. Most migraine attacks were moderate in severity (82.3%), associated with photophobia (75.8%) and phonophobia (72.7%), and resulted in some impairment of daily function (98.7%).
Efficacy
Based on the results of the interim analysis, which was conducted on 240 evaluable 12–17 year-old patients, the sample size for 12–17-year-olds was increased by the pre-specified amount of 100 patients. As a result of this change in the sample size, the conditional power was increased from 68.6% to 73.6% for the primary hypothesis. Efficacy results for the final analysis are summarized in Table 3. Results for the 2-hour pain freedom endpoint are illustrated in Figure 2. For the primary hypothesis, 2-hour pain freedom in 12–17-year-olds, rizatriptan demonstrated a statistically significantly higher response rate than placebo (30.6% versus 22.0%; p-value = 0.025). The pre-specified multiplicity strategy required statistical significance testing for each primary and secondary hypothesis to be performed in a specific order. The first of the three pre-specified secondary hypotheses, 2-hour pain relief in 12–17-year-olds, was not statistically significant. Therefore, the remaining two secondary hypotheses, 2-hour pain freedom in 6–17-year-olds and 2-hour pain relief in 6–17-year-olds, were precluded from formal statistical significance. However, in 6–17-year-olds, rizatriptan was nominally statistically superior to placebo (33.0% versus 24.2%; p-value = 0.010) for 2-hour pain freedom. In 6–11-year-olds, rizatriptan demonstrated a higher response rate than placebo (39.8% versus 30.4%) for the exploratory endpoint of 2-hour pain freedom, but this difference was not statistically significant (p-value = 0.269). The differences in response proportions between rizatriptan and placebo for 2-hour pain freedom were similar in 12–17 (8.6), 6–17 (8.8), and 6–11 (9.4) year-old populations. In a subgroup analysis by weight in the overall 6–17 year-old population, the percentages of patients with 2-hour pain freedom in those weighing <40 kg were 39.4% (39/99) for rizatriptan (5 mg) versus 36.7% (33/90) for placebo, whereas the percentages in those weighing ≥40 kg were 30.7% (87/283) for rizatriptan (10 mg) versus 20.5% (61/298) for placebo.
rMean percentage (95% confidence interval) of patients with pain freedom at 2 hours after Stage 2 dosing in: a) 12–17-year-olds (primary endpoint), b) 6–17-year-olds (secondary endpoint), c) 6–11-year-olds (exploratory endpoint). p-values are for comparison versus placebo. m indicates number of patients in the FAS population. Summary of efficacy by age group (full analysis set): endpoints are at 2 hours after Stage 2 dose except where stated. n: number of evaluable patients achieving endpoint (reported or carried forward); m: number of evaluable patients. Odds ratio and p-value computed using a logistic model adjusting for Stage 2 baseline pain severity (moderate versus severe) and region (US versus ex-US). An odds ratio > 1 favors rizatriptan. Rizatriptan group refers to 5 mg or 10 mg. p < 0.05, **p < 0.01; except for the primary endpoint, these symbols represent nominal p-values without adjustment for multiplicity. See text for the results of the sequential testing strategy to adjust for multiplicity that applied to the primary and secondary endpoints. Pre-specifed primary endpoint; bpre-specified secondary endpoint; all other comparisons were exploratory.

Although rizatriptan demonstrated a higher response rate than placebo for secondary endpoints of 2-hour pain relief in 12–17-year-olds (58.8% versus 51.4%; p-value = 0.080) and 6–17-year-olds (57.6% versus 52.6%; p-value = 0.178), the differences were not statistically significant. For the exploratory endpoint of 2-hour pain relief in 6–11-year-olds, there was not a significant difference between rizatriptan and placebo (54.1% versus 55.9%; p-value = 0.666).
The post hoc analysis for 2-hour pain relief using the alternative definition for the endpoint of a reduction of at least 2 grades from a baseline pain score of moderate/severe (Faces 3, 4, or 5) (13) demonstrated nominal statistical superiority of rizatriptan over placebo in 12–17-year-olds (51.8% versus 42.7%, respectively; p-value = 0.033) and 6–17-year-olds (52.4% versus 43.3%, respectively; p-value = 0.019). In 6–11-year-olds, rizatriptan had a higher response rate than placebo on this endpoint (54.1% versus 45.1%), but the difference was not statistically significant (p-value = 0.389).
The results also favored rizatriptan (based on nominal statistical significance) on many of the exploratory endpoints, including 2–24 hour and 2–48 hour sustained pain freedom, absence of nausea, and ability to function as usual in 12–17 and 6–17-year-olds, but not in 6–11-year-olds (Table 3).
No significant differences were seen between treatments in any of the age groups with regard to the percentage of patients who rated themselves as satisfied on the Overall 24-Hour Assessment of Study Medication Scale, although directionally the results favored rizatriptan. The percentages of patients who rated themselves as ‘a lot better’ for rizatriptan versus placebo were 33.8% versus 26.9% in 12–17-year-olds, 44.9% versus 29.7% in 6–11-year-olds, and 36.7% versus 27.6% in 6–17-year-olds.
Tolerability and safety
Summary of adverse events in within 14 days of dose by age group (all patients as treated).

As determined by the investigator to be related to the drug.
Triptan-related = asthenia, dizziness, dry mouth, fatigue, chest discomfort, throat tightness, myalgia, nausea, paraesthesia, somnolence.
Discussion
In this trial, rizatriptan was shown to be superior to placebo in the proportion of migraine patients 12–17 years of age reporting pain freedom at 2 hours, the protocol-specified primary outcome measure. Nominally significant differences from placebo were also observed for sustained pain freedom and sustained pain relief at 24 hours and 48 hours in 12–17-year-olds, and compared with placebo, rizatriptan appeared to reduce the incidence of associated symptoms of nausea and vomiting but not photophobia and phonophobia in 12–17-year-olds. Finally, a higher proportion of 12–17-year-olds on rizatriptan than placebo reported ‘as usual’ functioning after 24 hours.
These results support and extend the previous findings from Ahonen et al. (13) that a weight-based dosing regimen of rizatriptan was effective in relieving migraine pain in children and adolescents. Both studies used a 5-Face Pain Scale (14) rather than the 4-grade verbal scale typically used in adult studies (18) to allow patients more flexibility and options for expressing their pain experience. In the Ahonen et al. (13) study the primary endpoint was ‘pain relief’ at 2 hours, defined as a 2-grade (face) reduction on the 5-Face Pain Scale. For the present study, the endpoint of 2-hour pain freedom was used and 2-hour pain relief was a secondary endpoint pre-defined as a reduction in pain from ‘moderate or severe’ (Faces 3, 4 or 5) to ‘mild or none’ (Faces 1 or 2) on the 5-Face Pain Scale, analogous to the definition of pain relief derived from the standard 4-grade verbal scale in adult studies. Using this pre-specified definition of pain relief, rizatriptan was not significantly different from placebo in 12–17-year-olds. However, a post hoc analysis using the alternative Ahonen et al. (13) definition of pain relief did demonstrate a nominally significant difference.
Unlike the previous acute migraine treatment trials in children, the present study included a younger cohort of 6–11-year-olds who constituted approximately 25% of the total sample. As expected, owing to the limited target sample size and statistical power for comparisons in this age group (and therefore the absence of formal hypotheses related to 6–11-year-olds), differences on efficacy measures between rizatriptan and placebo were not statistically significant in this group. Inspection of the results, however, showed a numerical difference between treatment groups that appeared similar to that in the older children. Thus, when data from the 6–11-year-olds were combined with data from the 12–17-year-olds, the pattern of findings in the overall 6–17 year-old population was generally similar to that observed for the 12–17 year-old population.
An important element of this trial was the use of weight-based dosing. The patient populations defined by dose/weight aligned closely with those defined by age, which is why the sample sizes and results for the 5 mg dose group (those weighing <40 kg) were similar to those of the 6–11-year-old age group.
In the present study, the weight-based dosing regimen of rizatriptan was generally well tolerated across all age ranges for treatment of a single migraine attack. Only a minority (∼20%) of patients reported adverse events. Adverse events that did occur were generally similar to those observed in previous studies of rizatriptan in adults and adolescents and did not suggest serious safety concerns. There appeared to be a modest effect of dose, as adverse events were slightly more common in patients weighing ≥40 kg (who received rizatriptan 10 mg) versus those weighing <40 kg (who received 5 mg) and also in females versus males, although the overall pattern of adverse events was very similar across these subgroups and, importantly, were not markedly different from those observed in the placebo group.
Several previous studies of rizatriptan 5 mg in adolescent migraineurs failed to show efficacy (11,12). A number of factors could account for this, including potentially greater heterogeneity in the pediatric population and greater non-specific response. The present study incorporated several design elements in an attempt to control for some of these issues, including enrichment by selection of a population that had an unsatisfactory response to NSAIDs and acetaminophen, the requirement for patients to typically have untreated migraine durations of 3 hours or more (midway between the International Headache Society recommended durations of 2 hours for diagnosing migraine in children and 4 hours for diagnosing migraine in adults (17)), and the use of a placebo run-in. The favorable efficacy outcome of the present study and the previous Ahonen et al. study (13) using weight-based rizatriptan dosing also suggests that the failure to demonstrate efficacy of rizatriptan 5 mg in the previous adolescent studies (11,12) may have been due in part to underdosing.
Although the absolute response rates to rizatriptan were relatively high in the present study, response to placebo was also considerable, resulting in a modest overall treatment effect. Placebo response rates in pediatric migraine have typically been high. This study used a run-in enrichment design in an attempt to reduce the high placebo response rate of 56–69% observed for 2-hour pain relief in previous studies (11,12). Although the 15-minute run-in appears to have been effective in filtering out a number of ‘placebo responders’, response to placebo in this study, particularly for the 2-hour post-Stage-2 dose pain relief endpoint, was still high compared with acute migraine trials in adults (51% versus the 38% typically observed in previous adult rizatriptan studies (19)). It should be noted that response to placebo may not be consistent within an individual over time, so that identification of patients as ‘placebo responders’ in the 15-minute run-in of the present study does not imply that these patients would always be placebo responders.
Several factors limit the interpretation of these data. The study used an enrichment methodology and the results are thus relevant only to children whose symptoms do not respond to an initial intervention. Although the study used two different dose strengths, it did not explore dose response, as the different doses were assigned based on weight and the different dose groups are thus not comparable. Finally, there was no active comparator and we cannot, therefore, determine the efficacy of rizatriptan as compared with other treatment options for patients with pediatric migraine.
An important question, in light of the results of this study, relates to the role for rizatriptan in the treatment of pediatric migraine. The results demonstrate that rizatriptan is associated with a drug-specific effect on migraine pain in pediatric patients. However, the results also suggest that many pediatric patients with migraine will improve for non-specific reasons, and probably without pharmacologic intervention, suggesting that a conservative approach to initiating rizatriptan therapy is appropriate.
In summary, in this trial rizatriptan was more effective than placebo in eliminating pain and was generally well tolerated in the acute treatment of a migraine attack in pediatric migraineurs who had failed to respond to over-the-counter analgesics.
Footnotes
Funding
This study was funded by Merck & Co., Inc.
Acknowledgments
The authors would like to thank Nicole Dupre from Merck for assistance with study administration and Sheila Erespe from Merck for assistance in formatting the manuscript.
We were deeply saddened to hear of the death of our colleague and co-author Don Lewis shortly before this paper was submitted. Don was a champion for sufferers of many pediatric diseases, especially pediatric migraine, an area in which he published extensively. Among many other accomplishments, he led teams that created the first US national guidelines for evaluating and diagnosing children with recurring headaches (20) and US national guidelines for the pharmacological treatment of migraine in children (![]() ). Whenever you talked to Don you could feel his love and energy for his field and his commitment to advance treatments for children. The world of pediatrics has lost a great champion. He will be sorely missed.
). Whenever you talked to Don you could feel his love and energy for his field and his commitment to advance treatments for children. The world of pediatrics has lost a great champion. He will be sorely missed.
Elements of the data in this study were previously presented at the 53rd Annual Scientific Meeting of the American Headache Society, Washington DC, 2–4 June 2011, and the 15th Congress of the International Headache Society, Berlin, Germany, 23–26 June 2011.
External Data Monitoring Committee members
Guy McKhann MD, Michael Eliasziw PhD, Paul Graham Fisher MD, Gihan Tennekoon MD, Daphne T. Hsu MD.
Study investigators
Conflicts of interest
This study was funded by Merck Research Laboratories. TWH, KC, DM, YZ, CA, LHM, NS, RB, EM, CL, and DJH are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., and own or owned stock/stock options in Merck. EP, DL, and MH have received consulting fees from Merck.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.