
Abstract
Cytoskeletal dysfunction has been proposed during the last decade as one of the main mechanisms involved in the aetiology of several neurodegenerative diseases. Microtubules are basic elements of the cytoskeleton and the dysregulation of microtubule stability has been demonstrated to be causative for axonal transport impairment, synaptic contact degeneration, impaired neuronal function leading finally to neuronal loss. Several pathways are implicated in the microtubule assembly/disassembly process. Emerging evidence is focusing on Notch as a microtubule dynamics regulator. We demonstrated that activation of Notch signalling results in increased microtubule stability and changes in axonal morphology and branching. By contrast, Notch inhibition leads to an increase in cytoskeleton plasticity with intense neurite remodelling. Until now, several microtubule-binding compounds have been tested and the results have provided proof of concept that microtubule-binding agents or compounds with the ability to stabilize microtubules may have therapeutic potential for the treatment of Alzheimer’s disease and other neurodegenerative diseases. In this review, based on its key role in cytoskeletal dynamics modulation, we propose Notch as a new potential target for microtubule stabilization.
The Cytoskeleton and diseases
Cytoskeletal dysfunction has been proposed during the last decade as one of the main mechanisms involved in the aetiology of several neurodegenerative diseases [Bunker et al. 2004; Cairns et al. 2004; Cartelli et al. 2010].
Microtubules (MTs) are basic elements of the cytoskeleton composed of α- and β-tubulin heterodimers; the dysregulation of MT stability has been demonstrated to be causative for axonal transport impairment, synaptic contact degeneration, impaired neuronal function leading finally to neuronal loss. Several neurodegenerative diseases have been linked to impaired MT dynamics [Cappelletti et al. 2005] and axonal transport [Trojanowski et al. 2005; Roy et al. 2005]: hereditary spastic paraplegia [Rainier et al. 1998; Errico et al. 2002; Reid et al. 2002], familial motor neuron disease [LaMonte et al. 2002; Jablonka et al. 2004], Charcot-Marie-tooth disease type2A [Zhao et al. 2001; Tanabe and Takei, 2009], Huntington’s disease [Trushina et al. 2003], familial amyotropic lateral sclerosis [Julien et al. 2005; Mórotz et al. 2012], Parkinson’s disease and related synucleinopathies [Wersinger and Sidhu, 2005; Cartelli et al. 2012], Alzheimer’s disease [Terry, 1998; Kanaan et al. 2012], progressive supranuclear palsy [Morfini et al. 2002], frontotemporal dementias [Ittner et al. 2008] and related tauopathies. All of these pathologies are triggered by different events that finally converge on MT disruption/destabilization.
Neuron cytoskeleton has to be maintained in a condition of ‘dynamics balance’ between stabilization/destabilization to achieve the right degree of wellbeing. Indeed, equilibrium loss is linked to pathologic conditions. For example, in hereditary spastic paraplegia MTs are hyperstabilized, cytoskeletal structure is too rigid and neurons lack plasticity, essential for neurite branching and new connections formation [Fassier et al. 2013; Tarrade et al. 2006]; by contrast, in Alzheimer’s disease, MTs are too destabilized, axonal trafficking is impaired and synaptic contacts collapse [Zhang et al. 2012].
MTs extend in all directions throughout the cell, forming a dynamic network that continuously grows, retracts, bends and breaks. Therefore, rather than providing cellular rigidity, they are important for enabling dynamic processes such as intracellular transport or mitotic spindle formation that heavily depend on their ability to be polymerized, depolymerized and severed. The tight regulation of their dynamics is pivotal to ensure efficient transport of cargoes along the axons. With a severely destabilized MT network and a disturbed axonal transport system, neurons are not able to function properly and consequently degenerate [Almeida-Souza et al. 2011]. Loss of normal regulation of MT dynamics could have deleterious effects on cell viability; they can be considered as ‘biosensors’ of cellular wellbeing [Bunker et al. 2004].
Despite the diverse etiopathogenesis, the different neurodegenerative pathologies are linked to progressive accumulation of abnormal filamentous proteins; and this, together with the absence of other disease-specific neuropathological abnormalities, provides evidence implicating neuronal filaments and MTs in disease onset and progression [Cairns et al. 2004].
Microtubule-stabilizing compounds
Until now, several MT-binding compounds have been tested and the studies carried out have provided proof of concept that MT-binding agents or compounds with the ability to stabilize MTs may have therapeutic potential for the treatment of Alzheimer’s disease and other neurodegenerative diseases [Michaelis et al. 2002, 2005; Silva et al. 2011; Nelson, 2005; Butler et al. 2007].
Paclitaxel, a complex diterpene obtained from the Pacific yew (Taxus brevifolia), was the first natural product shown to stabilize MTs [Schiff et al. 1979]. It is able to prevent the disassembly of MTs and to promote their assembly. Paclitaxel (or Taxol, Bristol-Myers Squibb Company, New York City, USA) was originally studied and finally approved by the US Food and Drug Administration for clinical use in 1992 as a chemotherapeutic agent due to its ability to stabilize MTs in the mitotic spindle and arrest mitosis in cancer fast proliferating cells. In addition, it was demonstrated to have a beneficial role in an animal model of multiple sclerosis. Treatment with paclitaxel resulted in amelioration of clinical disease, reduction of gliosis and even remyelination [Moscarello et al. 2002].
It was subsequently studied in order to stabilize MTs of neuronal cells and to prevent axonal collapse and degeneration. To act as a neuroprotective compound, paclitaxel was used at doses far lower than those used in chemotherapy. It showed protective properties when used on neurons in vitro challenged with amyloid β [Michaelis et al. 2005] and it was able to enhance neurite outgrowth both in vitro and in vivo [Sengottuvel et al. 2011]. Paclitaxel also reduced deficits induced by mutant-tau transfection in cultured neurons [Shemesh and Spira, 2011], improved axonal transport rate, MTs number and motor function in a spinal cord tauopathy model [Zhang et al. 2005]. Unfortunately, in vivo studies demonstrated that it has a very poor entry in to the brain (less than 1% of the injected dose) [Moscarello et al. 2002], so the hypothesis of using paclitaxel for neuroprotection conflicts with the risk of peripheral accumulation and toxic side effects (neutropenia, unusual bruising or bleeding, gastrointestinal disease, fever, difficulty swallowing, ovarian damage and much more).
The same MT-stabilizing property emerged to be shared by other structurally complex natural products derived from microorganisms, plants and sponges. Among these, epothilone D (Epo D) displays interesting properties. Epo D derives from myxobacteria [Goodin et al. 2004] and besides being an efficacious MT-stabilizing agent, it is brain penetrant and has a good pharmacokinetics [Kolman, 2004; Andrieux et al. 2006; Brunden et al. 2010, 2012].
Another promising compound is NAP (generic name davunetide) (Allon Therapeutics Inc., Vancouver, BC, Canada), an MT protective agent [Gozes, 2011]. Gozes and colleagues tested NAP in several disease models and they found it to be a potent neuroprotective, memory-enhancing, neurotrophic agent and capable of inhibiting the aggregation of β-amyloid [Gozes et al. 2002; Ashur-Fabian et al. 2003; Matsuoka et al. 2007]. Surprisingly, even in human clinical trials, it was shown to increase memory scores in patients with amnestic mild cognitive impairment, a precursor of Alzheimer’s disease [Gozes et al. 2009] and to enhance functional daily behaviours in patients with schizophrenia [Javitt et al. 2012]. Furthermore, it has a good pharmacokinetics and can also be administered intranasally [Gozes et al. 2000, 2009].
Microtubule dynamics regulation
MT dynamics is spatially and temporally regulated by several pathways and MT-interacting proteins (see Table 1): tau, a MT-associated protein (MAP) that stabilizes MTs [Maccioni and Cambiazo, 1995], and katanin, the MT-severing enzyme [Roll-Mecak and McNally, 2010], both interact with the MT lattice; PAR-1 (also known as MARK) phosphorylates classical MAPs and detaches MAPs from MTs [Matenia and Mandelkow, 2009]; +TIPs, the MT plus-end-tracking proteins, specifically control the dynamic properties of the MT end [Gouveia and Akhmanova, 2010; Schuyler and Pellman, 2001]; ROCK pathway regulates MT dynamics via phosphorylation of the tubulin polymerization promoting protein 1 (TPPP1/p25) [Schofield et al. 2012]; (aPKC)-Aurora A-NDEL1 pathway is crucial for the regulation of MT organization during neurite extension [Mori et al. 2009]; Dishevelled (Dvl) pathway and the cooperation of Wnt-Dvl pathways increase MT stability though Gsk3β inhibition and c-Jun N-terminal kinase (JNK) activation [Ciani and Salinas, 2007]. These are only some of the known regulatory mechanisms that contribute to orchestrate MT dynamic remodelling.
Microtubule-interacting proteins.
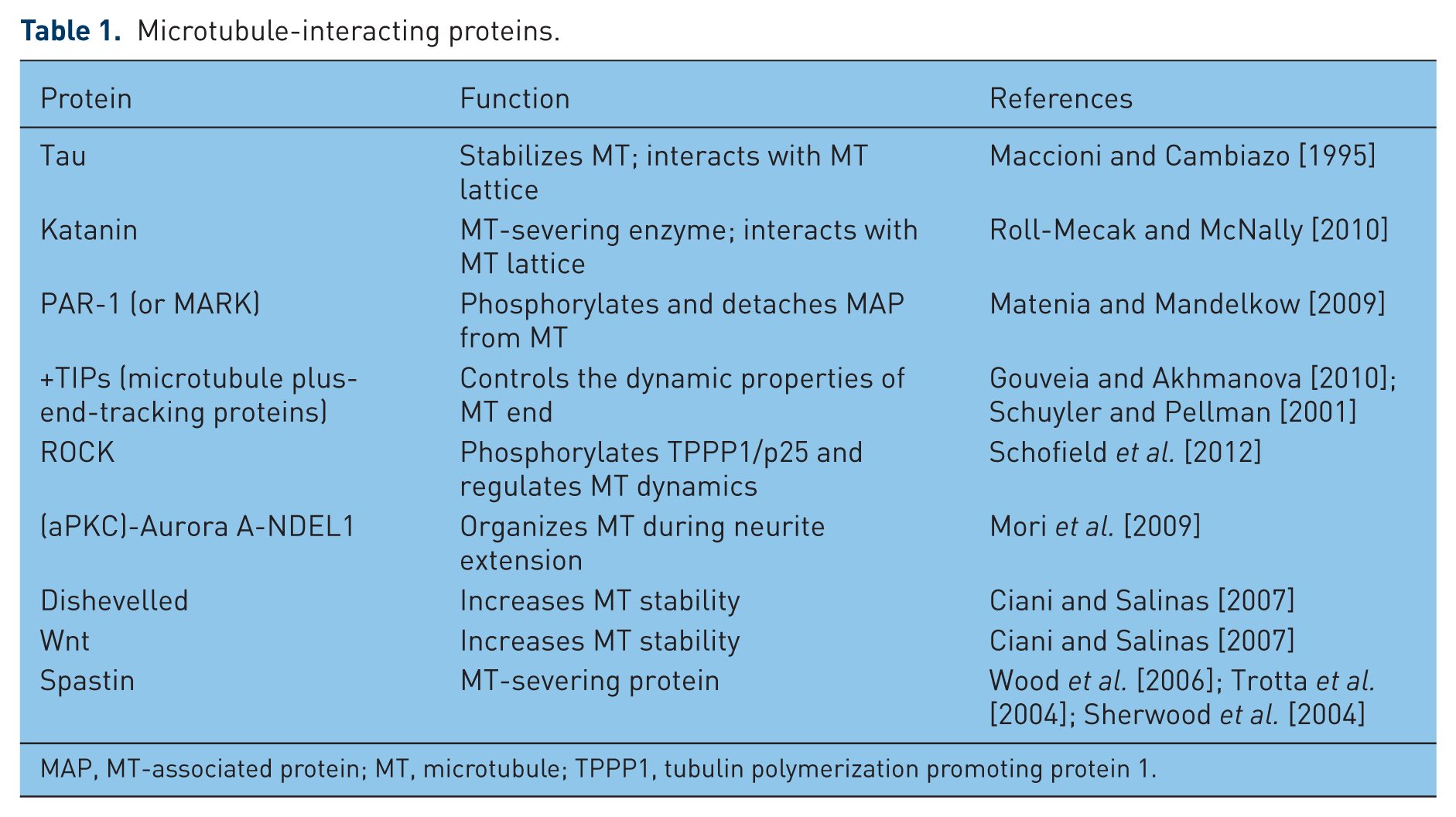
MAP, MT-associated protein; MT, microtubule; TPPP1, tubulin polymerization promoting protein 1.
Notch pathway
Notch pathway is recently emerged as a possible MT stability regulator. Notch is a heterodimer transmembrane receptor that, after binding with its ligands expressed on adjacent cells, goes through several proteolitic cleavages. The cytoplasmic cleavage by γ-secretase complex originates the active Notch intracellular domain; it translocates to the nucleus and triggers a transcriptional effects cascade. The best characterized target genes of Notch are the transcriptional repressors bHLH (basic helix loop helix) genes [the enhancer of split E(spl) complex in Drosophilia melanogaster and the hairy and enhancer of split (HES) and HES related (HESR/HEY) family genes in vertebrates].
In the past, Notch was considered a developmental protein that played a key role in cell fate decisions in uncommitted proliferative cells and in neurogenesis [Artavanis-Tsakonas and Simpson, 1991; Brennan et al. 1997; Go et al. 1998; Greenwald and Rubin, 1992; Hoppe and Greenspan, 1990]. In this context, Notch pathway activation results in inhibition of cellular differentiation and maintenance of a proliferative cellular pool [Louvi and Artavanis-Tsakonas, 2006]. For this reason, it started to be studied as a potential pharmacological target for several cancers [Nickoloff et al. 2003; Miele et al. 2006; Santos et al. 2006; Kunnimalaiyaan and Chen, 2007; Purow, 2012]. Finally, several Notch-targeting drugs (both inhibiting and activating, depending on the type of tumour) have been successfully chosen and tested in clinical trials for cancer therapies [Greenblatt et al. 2007; Fouladi et al. 2011; Sharma et al. 2012; Pinchot et al. 2011].
Notch role in postmitotic neurons
The initial suggestion that Notch signalling might have a role in postmitotic neurons came from the clear detection of Notch1 immunoreactivity in the nuclei of terminally differentiated postmitotic neurons in the rat retina [Ahmad et al. 1995]. A few years later, it was demonstrated that Notch1, apart from being highly expressed in embryonal mouse and human brain, continued to be expressed, although at lower levels, in the adult brain [Berezovska et al. 1998]. Furthermore, the cytoplasmic domain of endogenous Notch1 translocated to the nucleus during neuronal differentiation [Redmond et al. 2000].
Therefore, Notch activation could also occur in postmitotic or quiescent cells in the absence of division. A new role for Notch emerged in regulating developmental events subsequent to specification of cell fate. It was demonstrated that Notch is present on the growth cones of developing axons in Drosophila and it is required for axon guidance in both the central and peripheral nervous system [Giniger et al. 1993; Menne and Klämbt, 1994]. An involvement of Notch, as well as Reelin and Wnt, known to be active participants in neuronal maturation, was also suggested in neurodegenerative events underlying Alzheimer’s disease [Grilli et al. 2003; Lathia et al. 2008; Woo et al. 2009]. Notch expression resulted, markedly induced by excitotoxic stimuli in hippocampal pyramidal neurons [Ferrari-Toninelli et al. 2003]; this event resembles what happens at the onset of neurodegeneration in the adult brain, probably as an attempt to compensate neuronal loss by promoting neuronal growth. However, in aged brains, Notch1 signalling is reduced [Tanveer et al. 2012] and a chronic decrease in Notch1 function results in learning and memory deficits [Costa et al. 2003].
Research into Notch function in fully differentiated cells and in the adult brain was initially hampered because of the embryonic lethality of Notch knockout mice [Yoon and Gaiano, 2005]. Currently, with the development of Cre/loxP and viral gene transduction technologies, it is possible to manipulate Notch expression in mature animals, thus circumventing its developmental requirement [Han et al. 2002; Johnson et al. 2009; Ehm et al. 2010].
The first studies on the role of Notch in postmitotic neurons were carried out by using Notch antagonists or ligands to modulate the Notch signal in primary neuronal cultures. Notably, several ex vivo studies in different species clearly showed that modulation of the signal had a significant influence on neuronal morphology by affecting the extension of existing neurites (that is, axons and dendrites) [Sestan et al. 1999; Redmond et al. 2000; Berezovska et al. 1999; Qi et al. 1999].
Therefore, Notch plays a role in determining the only possible ‘cell fate’ decisions in postmitotic mature neurons: synaptic remodelling or neurite extension/retraction.
Notch as a microtubule stabilizer
The mechanism through which Notch can act on neurite morphology regulation is still a matter of debate. A proposal that has yet to be fully explored is that Notch is capable of influencing neuronal cytoskeleton [Giniger, 1998; Major and Irvine, 2005]. It has been suggested that the role of the Notch pathway in maintaining neuronal arborization in the adult is linked to its capability to modulate cytoskeleton plasticity [Louvi and Artavanis-Tsakonas, 2006].
We investigated the possible influence of the Notch pathway on MT stability and actually confirmed this hypothesis [Ferrari-Toninelli et al. 2008]. We demonstrated that activation of the Notch pathway in primary cortical neurons resulted in reduction of neurite branches and loss of varicosities. Varicosities appear as membrane swellings of various size and are regarded as presynaptic, dynamic structures that are able to remodel their morphology in response to a variety of stimuli [De Paola et al. 2003; Nikonenko et al. 2003; Udo et al. 2005; Umeda et al. 2005; Ferrari-Toninelli et al. 2009].
The changes in neurite morphology induced by Jagged1, a Notch ligand, were comparable to those induced by a low concentration of the MT-stabilizing drug paclitaxel. Evidence of increased MT stabilization, suggesting that the Notch pathway could also act through cytoskeletal modifications, was provided by the analysis of post-translational modification of tubulin. These modifications, which include detyrosination/tyrosination, polyglutamilation, polyglycilation, palmytoilation, phosphorylation and acetylation [Verhey and Gaertig, 2007], may occur individually or in combination. In this way, tubulin post-translational modifications play an important role in regulating MTs properties, such as stability and structure [Hammond et al. 2008]. Following Notch pathway activation, we found an upregulation of two post-translationally modified tubulins, the acetylated α tubulin and the polyglutamylated tubulin, commonly used as markers of stability [Hammond et al. 2008].
Notch downstream effectors
Several studies imply that Notch signals may modulate the cytoskeleton through local signal transduction pathways that do not require activation of nuclear gene expression. Different players have been identified to act together with or downstream of Notch. For example, Giniger showed that Notch interacts synergistically with the Abl tyrosine kinase to regulate the pathfinding of specific axons in Drosophila [Giniger, 1998]. Both Notch and Abl are present in the axon and the binding of Notch to Disabled (Dab), a protein that interacts with Abl, may explain how Notch communicates with Abl. Sanpodo is another possible mediator of neurite development regulation by Notch through its cytoskeletal interactions [Skeath and Doe, 1998; Dye et al. 1998].
The hypothesis of Notch-mediated neurite regulation by means of local modulators was interesting, especially considering activated Notch fragments that travel potentially great distances from growth cones to the nucleus. We explored this possibility, but our studies demonstrated that transcription and translation processes are necessary for the Notch-mediated morphological effect [Ferrari-Toninelli et al. 2009]. Alternatively, nuclear and local effects of Notch signalling may be integrated to regulate neurite development.
One of the links between Notch and a morphology-regulation-related protein was found by Hassan and colleagues: they reported a link with atonal, a proneural gene in the Drosophila nervous system. Characterization of atonal mutants indicated that in the brain atonal did not act as a proneural gene, but it was required for the proper axonal arborization of a subpopulation of neurons that innervate the optic lobe. Overexpression studies indicated that atonal and notch acted antagonistically in this population of neurons, with Atonal increasing axonal arborization and Notch decreasing it [Hassan et al. 2000].
Another possible player identified is neurogenin 3 (NGN3): Notch activation leads to expression of HES genes that inhibit NGN3 expression and finally reduces neurite outgrowth in the hippocampus. Therefore NGN3 acts to promote neurite outgrowth [Simon-Areces et al. 2010; Salama-Cohen et al. 2006].
In neocortical cells and in sensory neurons, Numb and numb-like (Numbl) are able to regulate axonal arborization acting as Notch antagonists [Huang et al. 2005].
We also identified a novel mechanism through which Notch is able to modulate neuronal cytoskeleton plasticity: by acting on the MT-severing protein Spastin. Stimulation of the Notch pathway by Jagged1 inhibited both the transcription and the expression levels of Spastin and induced MT stabilization and changes in axonal morphology [Ferrari-Toninelli et al. 2008]. Spastin gene mutation has been associated with axonal degeneration, leading to hereditary spastic paraplegia [Errico et al. 2002]. Further studies showed that Spastin is a MT-severing protein and its MT-destabilizing properties are fundamental for axon outgrowth and synaptic modulation in long motor neurons [Wood et al. 2006; Trotta et al. 2004; Sherwood et al. 2004]; interestingly, Yu and colleagues showed that in cultured neurons Spastin is more concentrated at the sites of branches formation and that protein downregulation resulted in neurite morphology changes, with a dramatic reduction of axonal branches [Yu et al. 2008]. Furthermore, it has been demonstrated that Spastin protein downregulation led to increased levels of acetylated and polyglutamylated tubulin, whereas Spastin overexpression caused a reduction of these post-translationally modified proteins [Trotta et al. 2004].
Therefore, we first established a link between the Notch signalling pathway and MT stabilization in postmitotic neurons, suggesting a novel endogenous pathway involved in modulating MT plasticity.
Notch microtubule-stabilizing effect is reversible
We found that Notch pathway activation acts as a MT stabilizer, and interestingly we demonstrated that this is a dynamic event that can be reversed. As evaluated by time lapse digital imaging, dynamic changes in cell morphology were rapidly reversible and dependent on the activation of the Notch signalling pathway [Ferrari-Toninelli et al. 2009]. Indeed, if Notch pathway activation causes MT stabilization followed by both neurite branching reduction and loss of varicosity, Notch pathway inhibition reverts such morphological effects and increases cytoskeletal plasticity. In particular, we observed morphological alterations (reduced neurite branching and loss of varicosities) in cortical neurons from transgenic mice characterized by Notch1 signalling hyperactivation (mice lacking the nuclear factor κB p50 subunit). The neuronal morphological effects found in p50–/– cortical cells were reversed after treatment with the γ-secretase inhibitor DAPT or Notch RNA interference [Bonini et al. 2011]. This means that, modulating the Notch pathway activation state, it is possible to act on MT dynamics hence cytoskeletal plasticity increasing or decreasing them depending on the necessity (see Figure 1).

Notch pathway effects on cytoskeletal plasticity.
Conclusion
In light of the growing evidence that MT dynamic balance maintenance could be beneficial in several neurodegenerative pathologies and even prevent them, we propose Notch pathway as a new possible pharmacological target for cytoskeletal protection.
Thinking about Notch as a possible target to protect the cytoskeleton presents several advantages. Notch can be modulated with activating or inhibiting compounds depending on the context and the effect required: Notch signalling activation results in increased MT stability and changes in axonal morphology and branching; Notch signalling inhibition leads to an increase in cytoskeleton plasticity with intense neurite remodelling. Some Notch-modulating compounds (such as the γ-secretase inhibitors) are already used in clinical trials for Alzheimer’s disease as β-amyloid reducing agents; it is well known that the Notch pathway and APP (Amyloid Precursor Protein) metabolism converge on γ-secretase proteolitic enzyme and that γ-secretase-inhibiting compounds, other than decreasing β-amyloid formation, also act on Notch pathway activation. Furthermore, it has recently been demonstrated that endogenous modulators of Notch signalling, as the endocannabinoid anandamide, can promote a shift in γ-secretase substrate processing to favour processing of Notch1 over APP, and this can confer neuroprotection [Tanveer et al. 2012].
Unfortunately, Notch may also present disadvantages: it is ubiquitously expressed, so the main challenge will be to find a compound able to pass the blood–brain barrier avoiding peripheral Notch side effects.
We suggest that a fine-tuned manipulation of Notch signalling may represent a novel approach to modulate neuronal cytoskeleton plasticity in order to guarantee neurons both the stability required to maintain their axonal architecture and the structural plasticity necessary to create new synaptic connections.
Finally, considering the relevant role of Notch in structural plasticity regulation and the importance of acting on MTs to protect neurons, we propose Notch as a new potential target for MT stabilization.
Footnotes
Funding
This work was supported by the Italian Ministry of Education, University and Research (Progetto di Ricerca di Rilevante Interesse Nazionale, PRIN 2009); and Fondo NEDD (Network-Enabled Drug Design), Accordi Istituzionali Regione Lombardia.
Conflict of interest statement
The authors declare that there is no conflict of interest.