
Abstract
Background:
Functional dyspepsia (FD) is common but few efficacious therapies exist. Itopride, a prokinetic, acts via dopamine D2 receptor antagonism and acetylcholinesterase inhibition, and is now approved in Japan, Mexico, and Europe for FD. However, long-term efficacy and safety data have not been published.
Objective:
To evaluate the long-term safety and potential effectiveness of itopride.
Design:
A long-term open-label drug effectiveness study.
Methods:
Males and females, 18–65 years, with FD (Rome II) and the absence (by upper endoscopy) of any relevant structural disease were recruited. After the double-blind treatment phase, patients were treated in an open-label extension phase.
Results:
A total of 798 patients were included in these two open-label trials and received at least one dose of study medication; 551 patients (69.0%) completed 6 months of treatment, and 294 patients (36.8%) completed a 12-month period. Response rates based on the global physician assessment were 61.7%, 64.8%, 69.0 %, 69.1%, 70.1%, 73.1%, and 77.9% at weeks 8, 16, 24, 32, 40, 48, and 52, respectively. Compliance was above 95%. The safety and tolerability profiles were as expected, with the majority of adverse events being gastrointestinal. Prolactin elevations occurred in 3% of the cases but were not clinically significant. No ECG changes were identified.
Conclusion:
In this population, itopride, given for up to 12 months, was safe, and up to two-thirds appeared to maintain symptom benefit.
Trial registration:
NCT00110968 and NCT00112203.
Introduction
The management of functional dyspepsia remains challenging.1,2 Functional dyspepsia (FD), defined by the Rome IV criteria as meal-related symptoms in the postprandial distress syndrome (PDS) subgroup and unexplained epigastric pain or burning in the epigastric pain syndrome subgroup, may affect as many as one in six in the general population. 3 Furthermore, approximately 5% of primary care practice deals with dyspepsia, of which the majority falls into the FD. 4 Following a normal endoscopy, treatment options for patients with FD include acid suppression therapy or Helicobacter pylori eradication, but the majority of patients fail to respond to both approaches.2,5
Prokinetic therapy has appeared promising in FD, although the efficacy of available therapies has been controversial. 6 In a meta-analysis, cisapride appeared to be superior to placebo, but concerns have been raised about possible publication bias, and cisapride has been withdrawn because of safety concerns. 7 In the same meta-analysis, the prokinetic tegaserod, a 5-HT4 agonist, was superior to placebo. However, the drug was recently withdrawn because of concerns about an increased incidence of cardiovascular ischemic events (0.1%). Despite continued off-label use in FD, the efficacy of metoclopramide or domperidone is not established in this condition.2,8 Acotiamide, a prokinetic that enhances the release of acetylcholine, has been approved in Japan and is effective for PDS symptoms with a favorable safety profile in multiple phase III studies.9,10
Itopride is a benzamide derivative that acts via dopamine D2 receptor antagonism and acetylcholinesterase inhibition. 11 In a dog model of pharmacologically induced gastric hypomotility, itopride restored motor activity in a dose-dependent manner. 12 A phase IIb multicenter, randomized, double-blind, placebo-controlled trial compared three doses of itopride (50, 100, and 200 mg with placebo) 13 ; in 548 Caucasian patients from Germany, there was a significantly higher proportion of responders based on a global efficacy measure versus placebo for all doses. A subsequent network meta-analysis concluded itopride was efficacious in FD. 8
Itopride is currently marketed in several countries including Japan, Singapore, Mexico, and Europe. Because it is highly polar, itopride does not cross the blood–brain barrier in any significant amount, and unlike cisapride it does not alter the Q-T interval. 14 However, the long-term safety and effectiveness of itopride in FD are unknown as there are no published follow-up studies. Two phase III clinical trials were conducted to assess the efficacy and safety of itopride given at a dose of 100 mg t.i.d (three times a day) in subjects with FD. 14 The global patient assessment responder rates at week 8 on itopride versus placebo were similar in both trials (45.2% vs 45.6% and 37.8 vs 35.4%, respectively). The patients included were then offered entry into an open-label, 12-month extension phase upon completion of the controlled trials. In addition, this open-label phase was also offered to “de novo” patients with a diagnosis of FD. Because itopride has now demonstrated efficacy in a recent meta-analysis 8 and itopride is becoming more widely available, the objective of the open-label trials reported here was to evaluate and report on the safety and real-world effectiveness of itopride HCl 100 mg three times a day when given for a period of up to 52 weeks. 8
Materials and methods
Two similar multinational, open-label trials were conducted. They took place in Germany, Belgium, the United States, and Canada.
Study population
Males and females, 18–65 years of age, were eligible for enrolment. All patients were required to sign an informed consent form before study evaluations.
De novo patients were required to have a diagnosis of FD by Rome II criteria prior to entry into the study.15,16 This was defined as the presence of persistent or recurrent dyspepsia (pain or discomfort centered in the upper abdomen). Discomfort could be characterized by or associated with upper abdominal fullness, early satiety, bloating, or nausea. Patients were required to have no evidence of organic disease (as confirmed by either an upper endoscopy during the past 6 months, conducted at the time of the screening visit) that was likely to explain the symptoms and no evidence that the symptoms were exclusively relieved by defecation or associated with a change in stool frequency or stool form, to exclude the irritable bowel syndrome.
Patients having completed the double-blind studies needed to have completed the 8-week trial to be eligible for the open-label study. Patients were not required to be symptomatic. A flowchart of the study has been included in the supplemental material of this manuscript.
Inclusion and exclusion criteria
For patients having completed the double-blind studies, they needed to have completed the double-blind study period; female patients had to have a negative serum pregnancy test and they all had to sign a new informed consent form prior to study entry. Patients with any newly occurring medical condition which was an exclusion for the double-blind phase were not eligible.
For patients not having been randomized to the double-blind study, they needed to be male or non-pregnant and non-lactating female outpatients between 18 and 65 years of age, with confirmed FD using the Rome II criteria and no evidence of organic disease that was likely to explain the symptoms. Concomitant H. pylori infection was permitted; patients suffering mainly or exclusively from symptoms corresponding to reflux disease were excluded.
Other exclusion criteria included patients with any clinically relevant ECG abnormalities, inflammatory bowel disease or celiac disease, any severe hepatic, renal, cardiac, metabolic, hematological, or malignant disease, and trimethylaminuria. Those who changed their smoking status, pregnant or lactating women, or those with a specific food intolerance that was relieved by changing their diet were excluded. Alcoholism or drug abuse and active psychiatric disease were also exclusions.
During this open-label phase, patients were required not to be on continuous nonsteroidal anti-inflammatory drugs (maximum of three consecutive days in a given week), anti-secretory medicine or any other treatment for FD.
Treatment
Patients having completed the double-blind phase kept the same number. De novo patients had a four-digit number assigned. These numbers were allocated in sequential order and registered in the patient enrolment list. Study medication was prepared in 21 tablet blister strips, packaged in a treatment unit box containing five strips. Two boxes were dispensed at each study visit. One tablet of study medication was taken three times daily: in the morning, at noon, and in the evening prior to meals. These two studies were conducted between September 2004 and September 2006. They were stopped by Axcan Pharma at that time, following a decision to discontinue this clinical program in light of the negative results obtained in the phase III program. 14
Compliance
At baseline and every 2 months, a sufficient supply of study medication was given. Pill counts were used to check compliance.
Outcome measures
Safety
Every 2 months, blood samples were collected and analyzed for safety. Adverse events and serious adverse event reporting, changes in concomitant medication intake, and compliance checks were performed. In addition, a physical examination and a 12-lead electrocardiogram were performed at months 6 and 12. For laboratory data, the normal range for prolactin levels was dependent on sex and was defined as 42–423 mIU/L for males and 42–613 mIU/L for females. A serum pregnancy test (if applicable) and urinalysis were performed at the start of treatment.
A listing of specified cardiac-related adverse events included the following: syncope, heart failure, angina, myocardial infarction, stroke, arrhythmias, palpitations, seizures, or ECG abnormalities.
Global patient assessment of efficacy
Every 2 months, the patient was asked to answer the question “Please rate the strength of your upper abdominal complaints in the past 14 days. Compared to the condition at the onset of treatment, how much have they changed? Please mark the statement that best applies to you: Symptom-free, markedly improved, slightly improved, unchanged, worse.” Symptom-free or markedly improved was defined a priori as a responder.
Patients who withdrew consent at any visit during the treatment period had to complete end-of-study procedures, that is a physical exam, ECG testing, laboratory evaluations (including serum pregnancy testing when applicable), concomitant medications recording, adverse event and serious adverse event reporting, and compliance check.
Post-study follow-up
For those patients with ongoing adverse events at the end of the study, concomitant medications were recorded, and adverse events were monitored and recorded.
Adverse events which were ongoing at study termination were followed for as long as necessary to adequately evaluate the patient’s safety.
Statistical plan
This study being conducted for safety purposes, a sample size calculation was not required. It was planned that at least 300 patients would be followed for a period of 24 weeks, and out of these, at least 100 patients were to continue treatment for another 28 weeks.
Data were analyzed by Quintiles Canada Inc.’s biostatistical staff. Summary statistics for safety and efficacy data collected during these open-label studies were presented in two groups:
Patients who took itopride during the double-blind studies (“Carry on”).
Patients who took placebo during the double-blind studies and “de novo” patients
All safety and efficacy assessments performed during the open-label studies were presented in analysis tables and data listings. Effectiveness was reported as the percentage of patients that significantly improved while taking the medication (completer analysis).
For qualitative parameters, the population size (N for sample size and n for available data) and the percentage (of available data) for each class of parameters were presented. Quantitative parameters were summarized by the population size (N for sample size and n for available data), the mean, the standard deviation, the median, the minimum, and the maximum values. All collected data were included in patient listings.
Drop-outs were not replaced during the study but were included in the data analysis to the extent that their data were available.
Classification into the safety population was conducted prior to database lock. Quintiles Canada Inc. generated the distribution list of patients to be included in the safety population before the database lock. Axcan Pharma Inc. had to approve this list before any analysis was performed.
Safety population
The safety population included all patients who received at least one dose of itopride during the open-label studies.
Handling of missing data
No imputations for missing Global Patient Assessment (GPA) of efficacy, laboratory data, vital sign data, medical history, or physical examination data were made.
Derived measures or scores were considered missing if the original measures required to calculate the derived measures or scores were missing. For example, if either a baseline or a post-baseline measure was missing for a particular patient, then a change from baseline to post-baseline was missing as well. Depending upon the measurement, analyses may not include all patients of an analysis population, because certain patients of an intended population may have missing data.
Baseline was defined as the latest evaluation performed prior to the first open-label dose of itopride 100 mg t.i.d. For patients enrolled in one of the two double-blind studies, the baseline refers to the end of study visit unless these patients were without treatment for more than 1 month between the end of the double-blind phase and the beginning of the open-label phase. In this case, a baseline visit was performed at the onset of the open-label study. For “de novo” patients, the baseline referred to the beginning of the open-label study.
If part of the date/time of an adverse event (AE) was missing, but the existing parts allowed determination of the timing of AE onset/end relative to the start and stop date of the first (open-label) itopride dose, then the AE was classified per review of the existing parts of the date/time fields. If the timing of the AE onset/end relative to the start and stop date of the first (open-label) itopride dose could not be made, then the AE was assumed to be a treatment-emergent adverse event (TEAE).
For non-serious AEs, if the intensity was missing, no imputation was performed. However, if intensity was missing for serious AEs, then it was classified in the most severe category in the summary tables.
If the assessment of the relationship between the AE and the study treatment was missing, it was classified as possibly related to the study treatment in the frequency tables of related treatment-emergent AE.
Results
Patient population
A total of 798 patients were included in the two clinical trials (Table 1). There were no major differences between the “de novo” subjects and the “carry-on” patients (Table 1). Patients enrolled in the study were on average aged 45 (“de novo”) and 43 (“carry-on”) years. Approximately two-thirds were female and over 90% were Caucasian. They had been diagnosed with FD around 52 months previously (“de novo”) or 39 months (“itopride carry-on”) before their enrollment. The timing of the onset of the first symptoms of FD was similar in the two groups (mean = 100.7 and 98.2 months, respectively), and about two-thirds had not taken any medication other than the study drug in the last 3 months.
Characteristics of patients and patient subgroups.
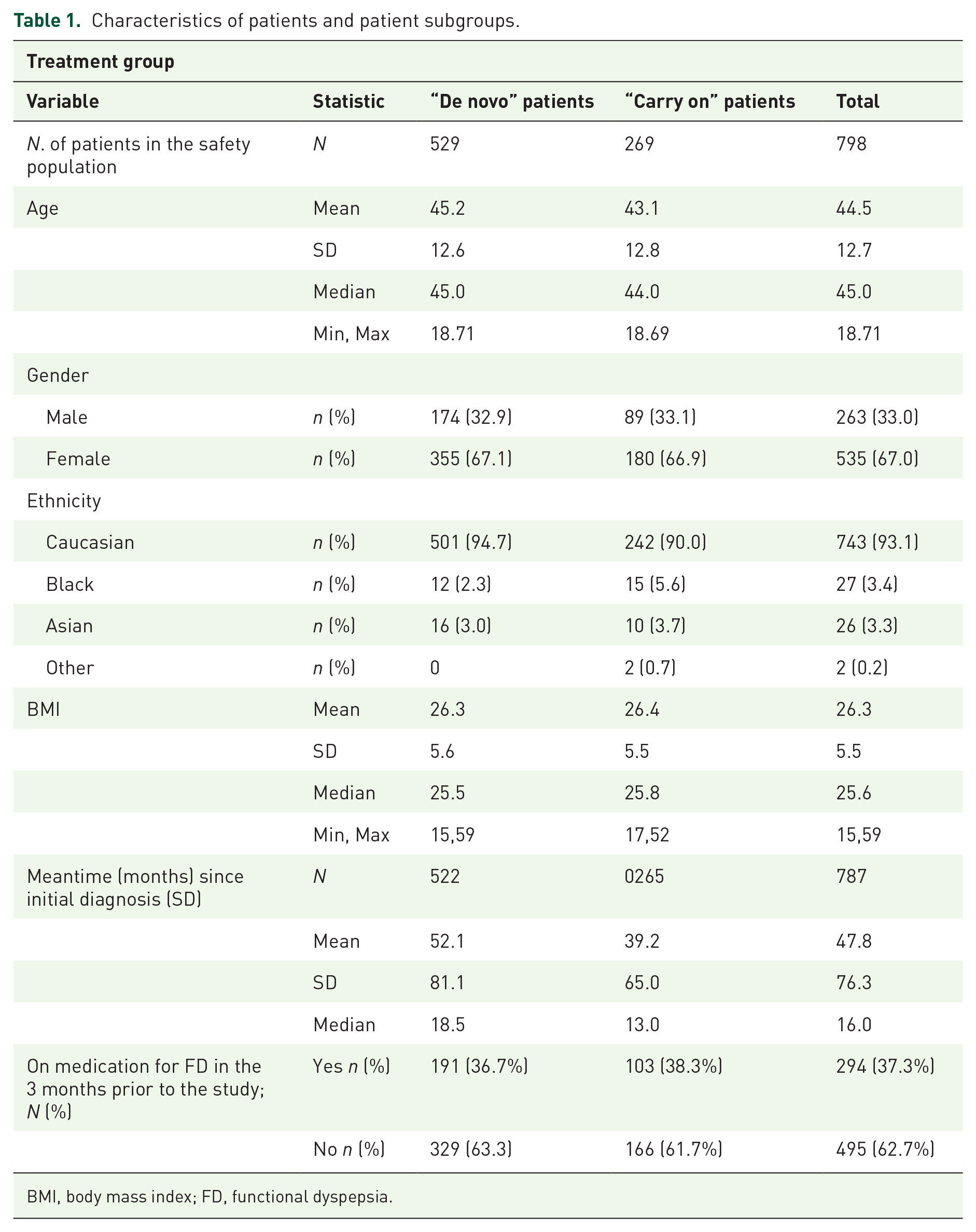
BMI, body mass index; FD, functional dyspepsia.
As illustrated in Table 2, the overall number of patients who completed the study was 421 patients (52.8%). The main reason for discontinuation was “other,” and that included mainly “Sponsor decision to stop the trial.” The number of patients who voluntarily withdrew their consent was 108 patients (25.5% of patients who withdrew or 13.5% of the overall study population).
Patient status at the end of the study (safety population).

Status missing on two subjects.
Percentages based on the number of patients who did not complete the study.
Mainly refers to the following reason “Sponsor decision to stop the trial.”
Effectiveness
The responder (symptom-free or markedly improved as defined a priori) rates based on the GPA of efficacy are detailed in Tables 3 and 4 for the entire patient population and each sub-group separately. In all subjects, response to treatment numerically increased with time, going from 61.7 % at week 8 to 77.9% at week 52. Note that this is a complete analysis of “Observed Cases” and not reported as a percentage of the intention to treat the population. At week 52, a total of 294 out of 798 (36.8%) were still on treatment.
Total responder rate (completer analysis) based on the GPA of efficacy: all patients.

Baseline values are for patients who completed the double-blind phase; includes both placebo and itopride groups.
Responder rates (completer analysis) for “de novo” versus Itopride “carry-on” patients.
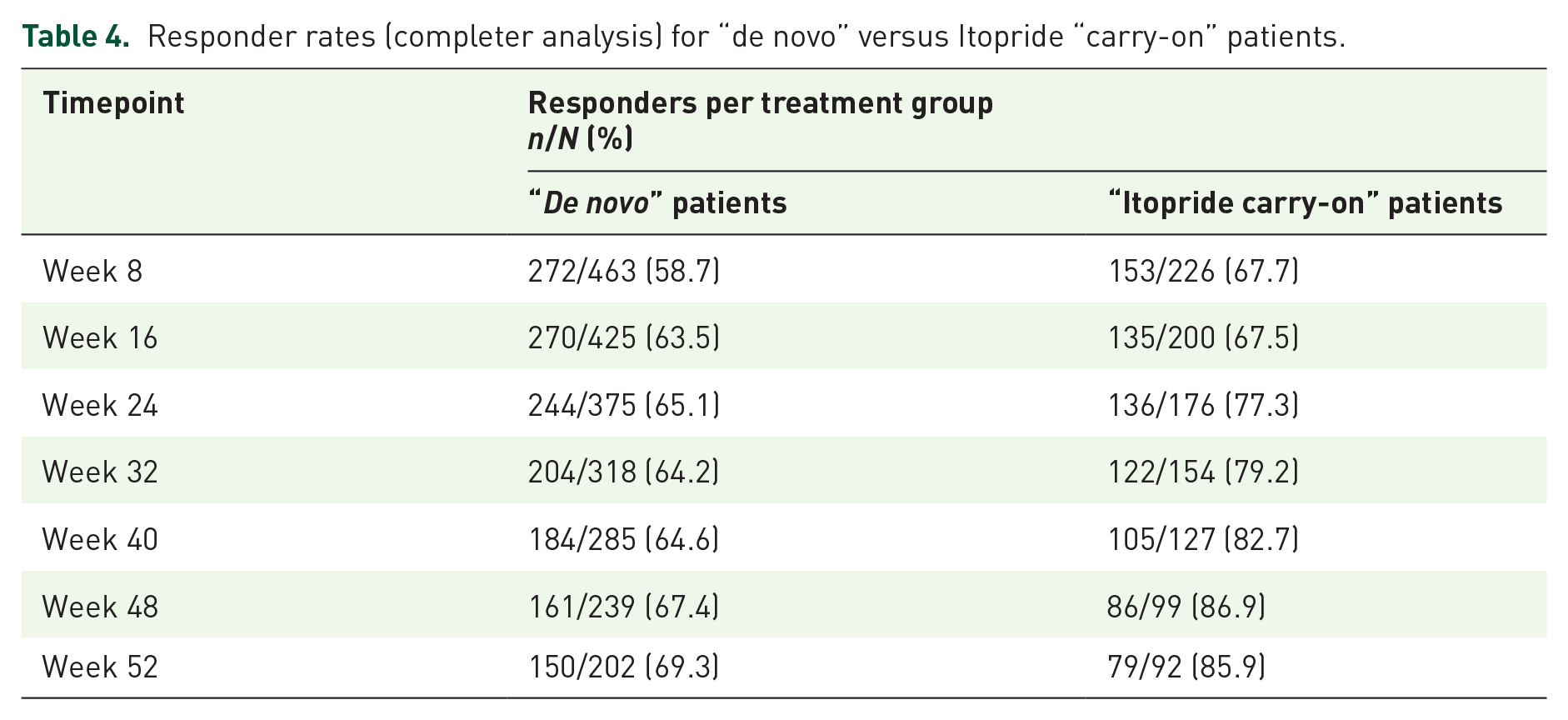
When looking at “de novo” patients and “carry-on,” the response rate also increased over time, with responder rates going from 58.7% at week 8 to 69.3% at week 52 for the “de novo” patients, while the “itopride carry-on” patients’ responder rate went from 67.7% at week 8 to 85.9% at week 52 (Table 4). The overall percentage of patients who were symptom-free increased from 12.8% (11.2%) for “de novo” and 15.9% for “itopride carry on”) at week 8 to 35.0% (31.2% for “de novo” and 43.5% for “itopride carry on”) at week 52. Drop-out rates were 43.6% and 40.7% in each respective subgroup. The proportion of patients discontinuing prematurely for lack of efficacy or treatment failure was numerically similar in both treatment groups (16.5% vs 18.8% of those who withdrew from the “de novo” and “itopride carry-on” populations, respectively, or 8.1% vs 11.1% from the total “de novo” and “itopride carry on” populations, respectively).
Overall, 73 patients withdrew due to lack of efficacy, accounting for 9.2% of the total study population.
Compliance, safety, and tolerability
Compliance to treatment was 96.1% and 97%, respectively, in each sub-group. Based on analyses of AEs, laboratory assessments, physical examinations, vital signs, and ECGs, itopride was generally safe and well tolerated, and the safety profile was similar to what has been observed in previous studies in subjects with FD.
The mean duration of exposure to itopride for the safety population was 265.3 days (n = 529) and 250.2 days (n = 269) for “de novo” versus “itopride carry-on” patients, respectively. A similar percentage of subjects in the “de novo” and “itopride carry-on” groups experienced TEAEs during exposure to itopride in these open-label trials (62.9% vs 59.1%, respectively). Individual TEAEs had similar incidences in the treatment groups, that is, the differences were below 2% for all TEAEs, except for urinary tract infection and headache. Treatment-emergent adverse events (TEAEs) occurring at an incidence ⩾1% in either group (n (%)) during the long-term extension phase study are listed in Table 5.
Incidence and type of adverse events.

Adverse events were coded using the MedDRA dictionary (Version 8.0). TEAEs are defined as any adverse event beginning on or after the study drug’s first dose up to 72 h following the last dose of the study drug. Patients experiencing the same TEAE multiple times are counted only once for the corresponding system organ class/preferred term. Percentages are based on the number of patients in the Safety population.
The vast majority of these TEAEs occurring at a frequency ⩾1% were mild to moderate in severity. The proportions of patients experiencing severe or study drug-related TEAEs were 4.0% and 15.8%, respectively. No differences were noted between the two treatment groups. Study drug-related TEAEs were defined as the events considered by the investigator as being possibly, probably or definitively related to study medication.
A total of 14 patients experienced 24 serious AEs, 3 of which were considered unlikely related to the study medication. The remaining 21 were considered by the investigator to be not related to the study drug. No subject died during the study. The proportion of patients discontinuing TEAEs was similar in both groups (7.8% vs 8.2% of the safety population for “de novo” and “itopride carry on,” respectively). The vast majority of TEAEs leading to discontinuation and considered related to study medication were gastrointestinal in nature.
Analysis of laboratory data revealed that itopride was associated with a mild to moderate increase in prolactin levels in a small proportion of patients. It occurred in 2.5% (13 patients) of the “de novo” and in 3.9% (10 patients) of the “itopride carry on” patients. This elevation in prolactin levels was clinically not significant but led to treatment discontinuation in three patients. Analysis of vital signs, 12-lead ECG, or physical findings did not reveal any clinically relevant effect of itopride including no effect on QT, QTcB, or QTcF.
Discussion
Following two randomized, double-blind, placebo-controlled trials, 14 we report the results of a long-term extension open-label safety and effectiveness study, enrolling close to 800 FD patients, of which 569 had participated in the phase III controlled trial program and 229 were newly enrolled patients. We found that itopride was well tolerated and safe, and over 70% of patients reported a sustained response, defined as being symptom-free or markedly improved. A placebo response of 37.5% has been reported in functional gastrointestinal disorders in a recent meta-analysis.13,17 Though not designed as an efficacy study, the responder rates observed in this long-term study were higher than the placebo response observed in the clinical trials (77.9%).
Itopride has been evaluated in an extensive phase II–III program. The phase II consisted of a large, placebo-controlled trial in FD in which 544 patients were randomly assigned to receive either itopride (50, 100, or 200 mg t.i.d) or placebo. 13 The co-primary outcome measures were the change in the Leeds Dyspepsia Questionnaire (LDQ) score relative to baseline after 8 weeks of treatment, GPA of efficacy every 2 weeks, and a composite response criterion with respect to the severity of pain in the upper abdomen and the severity of a feeling of excessive fullness after eating on the LDQ. The GPA demonstrated a response rate higher in the treated groups versus placebo (61.8% vs 40.7 % in the 100 mg t.i.d. dose vs placebo, p = 0.001) after 8 weeks of treatment. The response rate after 4 weeks of treatment was highest in patients treated with the 100 mg dose. The LDQ analyses revealed that itopride was significantly superior to placebo, with the greatest symptom improvement score in the 100 and 200 mg groups. In the LDQ, 18 the maximum total score is 40, and the higher the score the more severe the disease condition. In this phase II study, the total LDQ score decreased by 4.5 points in the placebo group, while the 100 and 200 mg doses decreased by 6.2 and 6.3 points, respectively. This translates into approximately a 25% improvement versus placebo which was statistically significant at the 0.05 level. Analysis of the combined endpoint of pain and fullness confirmed itopride superiority to placebo, with responder rates of 73% versus 63%, respectively (p = 0.04).
The phase III program was designed and conducted based on these observations. There were some differences in the patient population in phase III, notably as it relates to the presence of heartburn as a symptom. 16 Furthermore, to be classified as a responder this time a very strict definition was applied: patients had to be either symptom-free or markedly improved according to the global patient assessment of efficacy AND to have shown improvement with respect to the grading for severity of pain and fullness. The results were negative. 14
In parallel to those two pivotal phase III trials, a number of studies were conducted in an attempt to better understand the mechanism of action of itopride: (1) In a randomized study of healthy volunteers, itopride (100 mg and 200 mg doses) impaired gastric volume responses after a meal versus placebo, implying it may worsen gastric accommodation. 19 Despite this negative result, it is worth mentioning that the authors of this study argued that evidence of drug effects is best detected in patients with delayed gastric emptying and not in healthy volunteers with normal gastric function. (2) A cross-over study evaluating the effects of itopride 200 mg t.i.d. versus placebo on gastric emptying in longstanding diabetes was negative. 20 However, in that study, diabetic patients were not selected based on the presence or absence of gastrointestinal symptoms or gastroparesis. (3) Another study showed that itopride 50 mg or 100 mg t.i.d. inhibits transient lower esophageal sphincter relaxations without influencing esophageal motility, making it a potential drug for the treatment for gastroesophageal reflux disease. 21 In a randomized placebo-controlled trial from Europe of 100 patients with FD published in 2022, itopride significantly improved the dyspepsia score and all individual symptoms compared to baseline although the overall drug was not superior to placebo. 22 Notably, combining all the randomized trials of itopride in a network meta-analysis, the drug was superior to placebo in five randomized placebo-controlled trials studying 1854 patients, with a RR of 0.87. 8 We conclude from all the available data the patients most likely to respond to itopride do not have epigastric pain as a dominant feature, but this needs to be tested in future randomized trials.
In the long-term studies presented here, the GPA, which was a co-primary endpoint in the phase III controlled trials, was used to assess treatment efficacy at 8-week intervals (the LDQ was not measured to alleviate patient visits). Already after the first 8 weeks, the GPA responder rate was high and considerably higher than the rates reported during the controlled trials. 20 Throughout the treatment period, responder rates continued to rise to reach 78% by week 52. The Rome III committee suggests distinguishing between trials intended to demonstrate short-term versus long-term efficacy. 23 In this latter case, a 6-month trial duration is advised. 23 The sustained response rates in the present study might suggest itopride-related efficacy rather than a placebo or natural history effect. In a single tertiary care center longitudinal study by the Leuven group, it was reported that after a median follow-up time of 5 years, about 50% of FD patients reported improvement or resolution of symptoms. 24 In the absence of a placebo control arm in this study, it is not possible to distinguish true treatment response from spontaneous resolution of the symptoms over time in this study. On the other hand, the proportion of patients who were symptom-free in the present study increased from week 8 to week 52 by more than 20%, yet this strict outcome is usually associated with the lowest placebo response. 24 Nonetheless, the lack of a control arm makes it difficult to evaluate whether the sustained efficacy in the present study represents a placebo effect, a natural history effect, or the true efficacy of itopride over longer time use. Moreover, the high response rate may be partly attributed to the 9.2% dropout rate due to the lack of efficacy in the overall study population.
At every time point of evaluation, the GPA responder rate was higher in patients who had participated in the phase III trials, compared to the “de novo” enrolled patients. It is conceivable that this represents a recruitment bias, with FD patients experiencing a positive symptom evolution during the phase III study being more likely to be enrolled in the long-term extension phase. Similarly, patients who did not respond during the randomized phase may be less likely to enroll in the long-term extension phase. In our case, however, an almost equal number of responders and non-responders participated in the long-term study (46.4% vs 53.6%, respectively). Furthermore, 49.1% of non-responders were from the placebo group compared to 50.9% who continued from the itopride group.
A number of hypotheses could explain the apparent discrepant efficacy results in the phase II and open-label trials compared with the phase III placebo-controlled efficacy results. One could ask whether the dose was optimal. It is unclear if better results might have been obtained with a 200 mg dose. Another hypothesis could be a possible bias in the selected patient population. 22 It is possible that a subgroup of FD patients, for example only patients with postprandial non-painful symptoms, respond to itopride. Another aspect to consider is the choice of scales used to measure treatment efficacy, as different studies have used different primary outcomes. At the time of the trial initiation, the Rome III/IV recommendation 23 that the main outcome measure to be used in FD trials should be the proportion of patients achieving an a priori preset amount of improvement in symptoms did not yet exist. Furthermore, it is worth noting that even though the open-label studies included patients from Germany, Belgium, the United States, and Canada, the GPA was translated and verified across languages to ensure appropriate question interpretation.
Overall, itopride was well tolerated and compliance with the treatment was very high. Analysis of vital parameters and ECG showed no clinically relevant effects of itopride, including no significant effect on the QT intervals. In the original trials, 24-hour Holter monitoring was conducted on a subset of 30 patients with no significant changes identified in therapy. 16 Based on these data, itopride can probably be safely used in the general functional dyspepsia population, although it may be prudent to obtain a baseline ECG to exclude a prolonged QT interval when prescribing any prokinetic.
Elevation of prolactin occurred in 3% of patients on itopride but was not associated with clinical signs. Itopride is highly polar and thus is less likely to migrate through the blood–brain barrier compared to other prokinetic drugs or other dopamine antagonists. Radioactive studies in animals have shown that itopride distributes rapidly but no radioactivity was detected in the central nervous system. 25 Thus, any rises in prolactin levels are not likely to be pronounced as confirmed in our study and previous studies with itopride.13,14,26,27
No deaths occurred, and a total of 24 serious adverse events were registered during the trial. None of these events were considered related to the drug by the investigators. Treatment-emergent adverse events were reported for 60% of the patients. The only treatment-emergent adverse events with an incidence above 2% were gastrointestinal symptoms, as well as respiratory or urinary tract infections, headache, and back pain.
Limitations
This study is not free from limitations. First, this is an open-label trial without a control arm. As previously mentioned the lack of a control arm makes it difficult to evaluate whether the sustained efficacy in the present study represents a placebo effect, a natural history effect, or the true efficacy of itopride over longer-term use. Recruitment bias might have led to a selection of better responding patients although the study included responders and non-responders. Lastly, only patients seen at centers participating in the trial were included, and it is possible some patients were referred by other specialist care centers and not by primary care physicians.
Conclusion
In conclusion, in this long-term open-label treatment trial, itopride demonstrated effectiveness and a favorable safety profile during the 52-week treatment period, and all adverse events reported were mild and mainly gastrointestinal in nature.
Supplemental Material
sj-docx-1-tag-10.1177_17562848251321123 – Supplemental material for Itopride in functional dyspepsia: open-label, 1-year treatment follow-up of two multicenter, randomized, double-blind, placebo-controlled trials
Supplemental material, sj-docx-1-tag-10.1177_17562848251321123 for Itopride in functional dyspepsia: open-label, 1-year treatment follow-up of two multicenter, randomized, double-blind, placebo-controlled trials by Bert Broeders, Jan Tack and Nicholas J. Talley in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
We are grateful to all clinical investigators and study coordinators for having recruited patients in these clinical trials. As well, we would like to thank Mr. Rolland Gaudet (Quintiles Canada Inc.) for the statistical analysis and Dr. Monique Giguere, PhD Axcan Pharma Inc.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.