
Abstract
Background:
Pruritus is a symptom of several cholestatic liver diseases (CLDs) that can impair health-related quality of life (HRQoL). Despite evidence-based guideline therapy, managing cholestatic pruritus (CP) remains challenging, thus making the need for newer, more effective therapeutic agents more evident.
Objective:
Our study evaluated the efficacy of existing CP therapies.
Design:
Systematic review.
Data sources:
From inception until March 2023, we conducted a comprehensive search of MEDLINE, Cochrane, EMBASE, Scopus, ClinicalTrial.gov, and other sources, including pharmaceutical webpages and conference proceedings published in English that reported on CP interventions.
Methods:
Two reviewers independently conducted screening and full-text review of articles with extraction conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines. The methodological quality of studies included in our qualitative synthesis was assessed by using the Cochrane ROBINS-I and ROBINS-II tools for interventional studies and the National Heart, Lung, and Blood Institute Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies. The primary outcome assessed in our systematic review was the severity of CP after therapy.
Results:
Of 3293 screened articles, 92 studies were eligible for inclusion in the qualitative synthesis. Some patients’ HRQoL improved with evidence-based standard therapy. Others, particularly those with severe and refractory CP, often required conversion to or addition of experimental noninvasive (e.g., ondansetron) or extracorporeal liver support to alleviate CP. In addition, studies investigating a newer class drug, the ileal bile acid transporter inhibitor (IBATi), demonstrate its effectiveness in reducing serum bile acid and alleviating CP with sustained improvement noted in patients with the inherited childhood cholestatic disorders – progressive familial intrahepatic cholestasis and Alagille syndrome.
Conclusion:
Our findings consolidate data on the efficacy of guideline-based approaches and newer therapies for CP. While the initial findings are promising, additional clinical trials will be needed to determine the full extent of IBATi’s efficacy and potential use in treating other common CLDs. These results provide a foundation for future research and highlight the need for continued investigation into the management and treatment of CLDs.
Keywords
Background
The reduction or stoppage of bile flow characterizes cholestasis. It is a common complication of several chronic liver diseases, leading to elevated bilirubin levels that manifest as jaundice. Cholestatic liver diseases (CLDs) may arise from genetic, developmental, or environmental factors and can impact various stages of the hepatobiliary pathway. This includes (1) canalicular membrane dysfunction resulting in hepatocellular secretory failure, for example, intrahepatic cholestasis of pregnancy (ICP), progressive familial intrahepatic cholestasis (PFIC) occurring from protein mutations such as ATP8B encoded by FIC1 and bile salt export pump encoded by ABCB11 which corresponds to PFIC1 and PFIC2, and drug-induced cholestasis; (2) intrahepatic bile duct abnormalities, for example, primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and Alagille syndrome (AGLS); (3) extrahepatic obstruction caused by gallstones or benign strictures, for example, PSC, secondary sclerosing cholangitis, and tumor (cholangiocarcinoma, pancreatic carcinoma, and hilar lymph node metastasis). 1 Cholestasis affects several populations causing diverse symptoms, including jaundice. Approximately 80–100% of patients with cholestatic jaundice have pruritus, typically called cholestatic pruritus (CP). The prevalence of CP varies in the CLDs (see Table 1). CP can be mild and tolerable or severe and debilitating, resulting in sleep deprivation, fatigue, loss of concentration, and in extreme cases, suicidal ideation, which becomes a primary indication for liver (re)transplantation even in the absence of liver failure.2,3
Prevalence of pruritus in CLDs.

AIH, autoimmune hepatitis; ALD, alcoholic liver disease; ALGS, Alagille syndrome; ICP, intrahepatic cholestasis of pregnancy; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PBC, primary biliary cholangitis; PFIC, progressive familial intrahepatic cholestasis syndromes; PSC, primary sclerosing cholangitis; SSC, secondary sclerosing cholangitis.
Existing literature hypothesizes that in CLDs, potential pruritogens circulate systemically. When these molecules are excreted in bile, they undergo enterohepatic circulation (the efficient movement of bile acid molecules from the liver to the small intestine and back to the liver). As they undergo enterohepatic circulation, they are bio-transformed to pruritogens by cytochrome enzyme inducers in the liver and gut. The release of these pruritogens into plasma and tissues stimulates pruritogenic neural fibers in the skin that transmits an itch signal to the spinal cord and brain. 13 The most compelling evidence suggests that circulating serum bile acids (s-BAs), which activate a G protein-coupled receptor−MRGPRX4, are the principal pruritogens evident by CP attenuation following treatment with bile acid resins, plasmapheresis, apical sodium-dependent bile acid transporter inhibitors, nasobiliary drainage (NBD), etc. 6 Endogenous opioids have also been linked to CP pathogenesis. Its effect varies depending on the receptor subtype activated. Opioid activity on μ-opioid receptors (MOR) induces pruritus, while incitement of κ-opioid receptors (KOR) inhibits pruritus. An imbalance between MOR (increased) and KOR (decreased) may cause pruritus in conditions such as CLDs and end-stage renal disease. In addition, progressive liver diseases reduce the hepatic clearance of endogenous opioids, leading to increased circulation of endogenous opioids. Lysophosphatidic acid (LPA), a signaling molecule that acts on the G-coupled receptor (LPAR5) of many cells, autotaxin (ATX)−an enzyme that hydrolyzes lysophosphatidylcholine into LPA, serotonin, Substance P acting on NK-1 receptors, and histamine on H1 and H4 receptors have also been linked to pruritogenic signaling cascade in CLDs.4,14–16 Because the underlying mechanism of CP has yet to be fully understood, managing CP remains challenging.
The initial approach to CP involves addresses the underlying cause, such as endoscopic treatment for dominant strictures in PSC and discontinuing causative medications in drug-induced cholestasis. Mild CP can be relieved with warm baths, emollients, and antihistamines. For moderate to severe cases or when the underlying pathology cannot be treated, the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver suggest a stepwise drug approach.16,17 However, the effectiveness of this stepwise drug approach varies, working for some patients but not others.1,18 As a result, there is growing interest in exploring experimental methods for CP treatment. Our systematic review aims to provide an up-to-date and comprehensive review of conventional and investigational approaches to managing CP, which would be helpful for clinicians, researchers, and patients.
Data sources and methods
Search strategy
We comprehensively searched four major databases: EMBASE, Ovid MEDLINE, Scopus, and Cochrane Library. From their inception until 11 March 2023, we searched these databases to identify all relevant studies on CP therapies. We also explored ongoing clinical trials related to CP therapies on ClinicalTrial.gov and reviewed conference proceedings. This was necessary because CP therapies have rapidly evolved in recent years, and new therapies may have yet to be fully documented in published literature. To ensure the accuracy and completeness of our review, we followed the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines, a widely recognized tool for conducting systematic reviews. We limited our search to articles published in English to ensure consistency in our review.19,20
To capture the most relevant studies, we used a set of keywords that included ‘pruritus’, ‘cholestasis’, ‘cholestatic pruritus’, ‘cholestasis associated pruritus’, ‘cholestatic liver disease’, ‘itch’, and ‘IBAT inhibitors’ in our initial review.
Study eligibility
The methods used in this study involved conducting a comprehensive literature search across various databases, with no limitations placed on the duration of the studies or the follow-up period. The search results were then screened by two independent reviewers using a two-step process, which involved evaluating the title and abstract of each article, reviewing the full text of relevant articles, and examining the references cited within those articles to identify any additional relevant publications. Inclusion criteria: (a) experimental and observational studies related to CP therapy that reported our study’s primary endpoint, that is, the severity of CP after systemic treatments. Exclusion criteria: (a) non-human subjects’ studies; (b) studies not published in English; (c) studies that did not investigate CP, letters, and systemic review articles; and (d) studies that did not provide sufficient data on participants’ responses before and after CP treatment. To ensure the reliability of the study, any discrepancies between reviewers were resolved by consensus. This process helped to ensure that the final sample of articles included in the analysis was relevant and met the study’s inclusion criteria (see Figure 1).
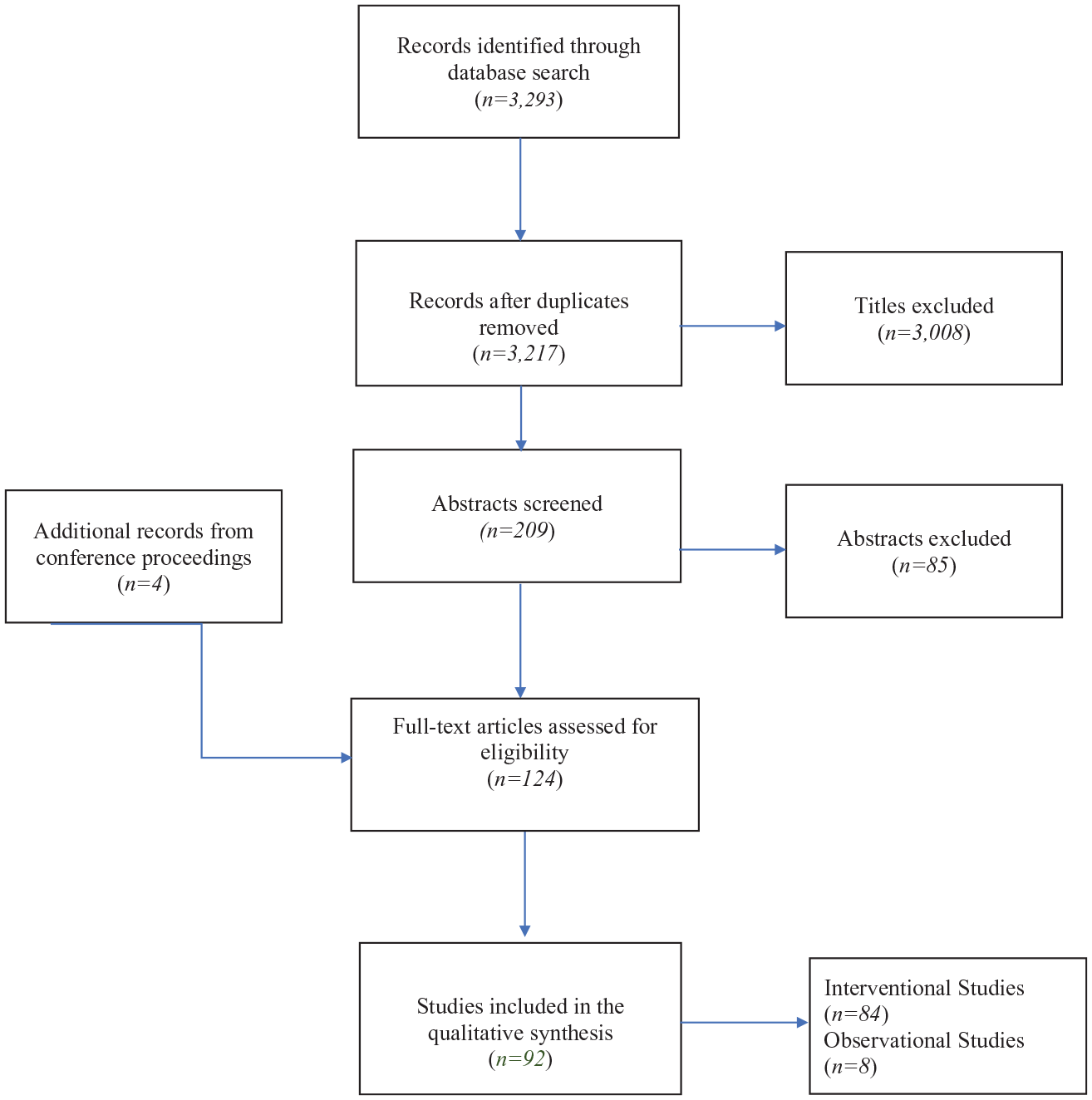
PRISMA flow diagram of study selection.
Risk-of-bias assessment
To evaluate the methodological quality of the experimental studies included in our systematic review, we used the Cochrane risk-of-bias tool for randomized trials (ROBINS-II) and non-randomized studies – of interventions (ROBINS-I). This assessment was carried out independently and was based on several parameters. These included the sequence generation for the randomization of subjects, allocation concealment, blinding of participants and personnel, blinding of outcome assessor, attrition bias, selective reporting, and other sources of bias. Clinical trials that did not meet any of the first three parameters were considered to have a high risk of bias. 21 We also used the National Heart, Lung, and Blood Institute Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies to provide an overall quality rating for observational studies included in our qualitative synthesis. A high risk of bias translated to a rating of poor quality. Low risk of bias translated to a rating of good quality. 22
Outcome measures
In our study, we aimed to determine the efficacy of systemic treatments in reducing the severity of CP. Due to its complex nature involving sensory and emotional components, pruritus is regarded as a multidimensional construct and has been evaluated using a variety of scales and tools. These include validated measures such as the Visual Analog Scale (VAS) and the Numerical Rating Scale (NRS) which assess the intensity of itch, as well as the Itch Questionnaire that examines different dimensions of pruritus such as its duration, frequency, location, impact on quality of life, and the Patient-Oriented Scoring Itch Assessment (POSITA) that explores the subjective experience of pruritus, including its sensation, urge to scratch, and impact on daily activities. Other methods include the composite peak VAS (CP-VAS) and non-validated scales determined by clinicians or patients. Previous research indicates that the minimal clinically important difference for improvement in pruritus, as measured by VAS and NRS, is a reduction of 2–3 points from the baseline.23,24 Our study’s secondary objectives included assessing the impact of systemic treatments on liver biochemistries, such as s-BA levels and drug safety. Statistical significance was set at a p-value of <0.05. 25
Results
Literature search
Our search yielded 3293 unique titles containing our specific keywords. After the removal of duplicates, 3217 records were screened. Of these, 3008 studies were excluded. Of the remaining 209 articles screened, we retained 124 based on the title and abstract. Ninety-two studies were ultimately included in the qualitative synthesis – 84 interventional and 8 observational studies (see Figure 1).
Risk-of-bias assessment results
The risk-of-bias assessment for the interventional and observational studies is reported in Supplemental Tables 1 to 3.
Therapeutic guidelines for the management of pruritus associated with cholestasis
An evidenced-based stepwise or experimental approach may be warranted in the management of CP (see Table 2).
Treatment strategy for managing CP.
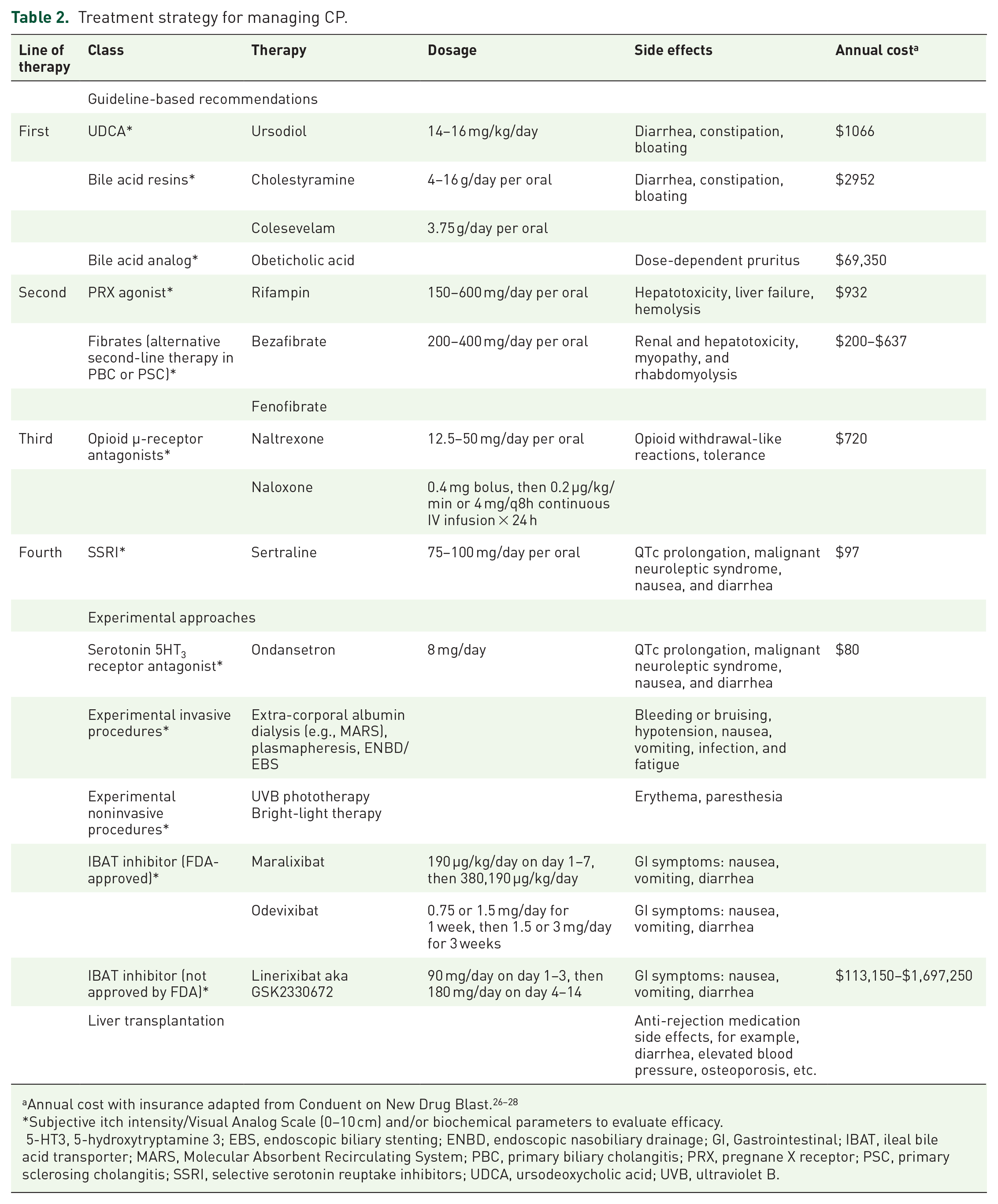
Subjective itch intensity/Visual Analog Scale (0–10 cm) and/or biochemical parameters to evaluate efficacy.
5-HT3, 5-hydroxytryptamine 3; EBS, endoscopic biliary stenting; ENBD, endoscopic nasobiliary drainage; GI, Gastrointestinal; IBAT, ileal bile acid transporter; MARS, Molecular Absorbent Recirculating System; PBC, primary biliary cholangitis; PRX, pregnane X receptor; PSC, primary sclerosing cholangitis; SSRI, selective serotonin reuptake inhibitors; UDCA, ursodeoxycholic acid; UVB, ultraviolet B.
Ursodeoxycholic acid is the primary treatment option for patients with CP related to ICP and PBC
Ursodeoxycholic acid (UDCA) is the recommended first-line therapy for CP associated with PBC and ICP. 23
Studies on the efficacy of UDCA in ICP
A randomized clinical trial (RCT) evaluated the efficacy and safety of UDCA 8.7 mg/kg/day compared with cholestyramine 8 g daily in ICP. The primary endpoint was a reduction of pruritus by >50% after 14 days of treatment. Pruritus intensity was quantified daily by patients using a score from 0 to 4 (0, no pruritus; 1, occasional; 2, intermittent pruritus daily with prevailing asymptomatic periods; 3, intermittent pruritus every day with symptomatic periods prevailing; 4, constant pruritus day and night). Secondary endpoints included s-BA levels and drug safety. In the study, UDCA effectively reduced pruritus compared to cholestyramine (66.6% versus 19.0%; p < 0.005), and endogenous s-BA levels decreased by 59.5% and 19.0%, respectively (p < 0.02). 29
In another RCT on the efficacy of UDCA 600 mg/day therapy in ICP, the severity of pruritus was measured before treatment and at day 20 with a pre-specified pruritus score system (0, absence; 1, occasional; 2, discontinuous pruritus with prevailing asymptomatic lapses; 3, discontinuous pruritus with prevailing symptomatic lapses; 4, persistent pruritus day and night). The study reported a significant reduction in pruritus score [1.50 ± 0.26 versus 3.25 ± 0.36 (baseline); p < 0.001) and serum bile salt levels (10.17 ± 2.80 versus 42.16 ± 11.05 (baseline); p < 0.001]. 30
In a third randomized trial, patients with ICP received either 450 mg/day UDCA or a placebo for 14 days during the third trimester of pregnancy. The severity of pruritus was registered, and VAS scores (VASs) were measured pretreatment and then weekly. The UDCA group reported a significant improvement in VASs (p = 0.007). 23
A fourth RCT assessed the effectiveness of UDCA in ICP. Participants were given either 500 mg of UDCA or a placebo twice daily (BID). The dosage was adjusted as needed until delivery or until the participant received early-term delivery or expectant management. The primary outcome assessed for UDCA was a maternal itch, measured using a 100 mm VAS of the worst itch experienced in the past 24 h, and whether early delivery increases the incidence of cesarean delivery. The results showed that UDCA reduced itching by 16 mm [95% confidence interval (CI): −27 mm to −6 mm], falling short of the pre−specified 30 mm difference by clinicians and patients considered clinically meaningful. Early-term delivery did not increase the incidence of cesarean delivery, with 7 out of 30 (23%) in the early-term delivery group and 11 out of 32 (33%) in the expectant management group undergoing cesarean delivery (relative risk 0.70, 95% CI: 0.31–1.57) 24
A fifth RCT compared UDCA 450 mg/day to S-adenosyl-L-methionine (SAMe) (1000 mg/day intramuscularly) given for ⩾15 days to patients with ICP. The severity of pruritus was graded pretreatment and subsequently, every 3 days using a pre-fixed pruritus score (0, absence; 1, occasional; 2, discontinuous pruritus during the day, with prevailing symptomatic relapses at night; 3, permanent pruritus day and night). All patients treated with UDCA showed a complete resolution of pruritus within 3 days, while no patient treated with SAMe had complete regression of pruritus. UDCA was determined superior to SAMe at lowering s-BA levels (p < 0.02). 25
Another RCT evaluated the efficacy of UDCA 500 mg twice daily with increased or decreased dosing in gestational cholestasis (min: 250 mg/day, max: 2000 mg/day). The primary outcome was a composite of perinatal death, preterm delivery, or neonatal unit admission for at least 4 h. Secondary outcomes measured included maternal itch, s-BA levels, and alanine transaminase levels. The study showed that UDCA does not reduce adverse perinatal outcomes in gestational cholestasis. However, the maternal itch score was lower in the UDCA group than in the placebo group [mean difference −5.7 mm (95% CI: −9.7 to −1.7)]. The reduction in the s-BA level was smaller in the UDCA group compared with the placebo group [adjusted geometric mean ratio 1.18 (95% CI: 1.02–1.36), p = 0·030]. By contrast, a reduction in alanine transaminase concentration was found in the UDCA group compared with the placebo group [adjusted geometric mean ratio 0.74 (0.66–0.83), p < 0·0001]. 31
Another randomized, controlled, open-label trial at multiple centers compared the effectiveness of UDCA and SAMe as monotherapy and in combination for treating ICP. Patients were randomly assigned to receive either UDCA 4 × 250 mg/day (Group 1), IV SAMe 1000 mg/day (Group 2), or a combination of both drugs (Group 3) until delivery. All treatments resulted in a significant and equal improvement in pruritus. Moreover, in all groups, there was a significant decrease in the s-BA levels, transaminases, and bilirubin after treatment (p < 0.05). Group 1 was found to be more effective than Group 2 in reducing s-BA levels (p < 0.05), and both Group 1 and Group 3 were more effective than Group 2 in reducing transaminases (p < 0.05). Additionally, Group 1 was associated with a higher likelihood of delivering at term. No perinatal deaths or adverse drug reactions were observed. 32
In contrast, an RCT using a semiquantitative scale of grades 1–4 did not find UDCA 300 mg BID nor SAMe 500 mg BID superior to the other in alleviating pruritus caused by gestational cholestasis. UDCA was only more effective at improving the concentration of the s-BA level and other tests of the liver function. 33
Studies on the efficacy of UDCA in PBC
An RCT was conducted to compare the effectiveness of UDCA 500 mg daily to a placebo in individuals with PBC. The study showed a significant decrease in itching severity, as reported by patients, and decreased consumption of cholestyramine, as recorded in a diary, in both groups (p < 0.01). The UDCA group had a significant reduction in abnormal serum bilirubin levels compared to the placebo group (p < 0.05). 34
A randomized open-label trial evaluated the effectiveness of UDCA in patients with PBC and refractory itching. Participants received UDCA alone (600 mg once daily) for 8 weeks or an equivalent dose of UDCA for 4 weeks, followed by UDCA combined with colestilan (CLL) (600 mg plus 6.42 g once daily) for another 4 weeks . Participants graded their itching intensity using a self-administered questionnaire and a numeric scale at baseline and 2-week intervals (0−10, 1 meaning no itching, and 10 indicating severe and constant itching). The study showed no significant change in itching score in the UDCA monotherapy group (2.5 ± 0.3 at week 8 versus baseline 3.7 ± 0.3, p > 0.05). Furthermore, the change in itching score was not significant at week 4 of UDCA alone (3.8 ± 0.4, p > 0.05) compared to the substantial improvement in itching score at week 8 of UDCA plus CLL [1.2 ± 0.2 at week 8 versus 4.0 ± 0.3 (baseline), p < 0.05]. 35
Another RCT assessed the effectiveness and tolerability of UCDA (13−15 mg/kg/day) in PBC. The primary endpoint was treatment failure − defined as a doubling of the bilirubin level to more than 70 µmol, the occurrence of a severe complication of cirrhosis, or a side effect requiring therapy interruption. The study showed that itching resolved in 40% of the patients in the UDCA group versus 19% in the placebo group, and the relative risk of treatment failure was three times higher in the placebo group. 36
In another RCT that evaluated the long-term effects of UDCA (14–16 mg/kg/day) in PBC, a significant reduction in itching score was observed in the UDCA group [1.5 ± 0.1 versus 2.0 ± 0.1 (baseline); p < 0.001] compared to placebo [1.7 ± 0.1 versus 2.0 ± 0.1 (baseline); p < 0.05]. At the same time, there was a progression of the histological stage, ductular proliferation, and ductopenia in patients that received a placebo. In addition, comparing the liver biopsy at the study entry and the end, UDCA demonstrated improved portal inflammation and prevention of histological stage progression. 37
Another intervention study on the long-term effects of UDCA 600 mg three times daily (TID) in PBC patients observed that in seven of eight patients with itching, itching disappeared a month after UDCA administration. In addition, portal inflammation activity decreased in all five patients who had undergone follow-up liver biopsies more than 1 year after UDCA administration. 38
An RCT also assessed the effectiveness of tauroursodeoxycholic acid (TUDCA), a metabolite of UDCA, in individuals with chronic hepatitis and pruritus, using daily doses of either 500 or 750 mg. Pretreatment: the VASs were 0.13 ± 0.35 for the control group and 1.18 ± 1.24 for the treatment group. After 6 months, a decrease in pruritus by 30% was observed in the TUDCA group, while an increase in pruritus by 50% was observed in the placebo group (p < 0.05). 39 Common side effects of UDCA or TUDCA included gastrointestinal symptoms like abdominal pain, flatulence, and diarrhea.31,32,37
Bile acid sequestrants are the preferred initial treatment for CP that is not associated with PBC or ICP
Cholestyramine and colestipol are two types of bile acid sequestrants used as first-line therapy for moderate to severe CP caused by conditions other than PBC and ICP. These medications bind to bile acids in the small intestine, preventing reabsorption and promoting excretion through feces.
Studies on the efficacy of bile acid sequestrants
Clinical trials have investigated the efficacy of bile acid sequestrants in managing cholestasis symptoms, particularly pruritus (itching) and s-BA levels. For example, one RCT found that cholestyramine (3 g TID) significantly reduced s-BA levels and itching scores in patients with intra- and extrahepatic cholestasis (VAS, 0–100 mm: −63.6%, p < 0.01) compared to placebo (VAS, 0–100 mm: +24.7%, p < 0.01) at week 4. In addition, the itching intensity increased in the placebo group, with a positive linear relationship between itching and s-BA levels found (p < 0.01). 40
Another RCT examined the efficacy of colesevelam (1875 mg BID) in patients with cholestasis of multiple etiology, including PBC and PSC. The primary endpoint was the proportion of patients with at least a 40% reduction in VASs. At week 3, 36% of patients in the colesevelam group reached the primary endpoint versus 35% in the placebo group, but neither was superior in reducing pruritus score (p = 1.0). By contrast, the mean s-BA levels that were comparable between the groups at baseline (p = 0.74) were significantly different post-treatment (p = 0.01) in favor of patients treated with colesevelam. Reported side effects of bile acid sequestrants are primarily gastrointestinal, including diarrhea and bloating. Therefore, patients should be monitored for these adverse effects during treatment. 41
Alternate treatments for patients with CLDs who have not fully responded to or cannot tolerate UDCA
A. Obeticholic acid
The FDA-approved obeticholic acid (OCA) is a potent nuclear hormone farnesoid X receptor (FXR) ligand. Activation of FXR by OCA inhibits bile acid synthesis, thereby protecting against toxic bile accumulation and facilitating hepatic regeneration. OCA is combined with UDCA or as a monotherapy when UDCA is not tolerated. The use of OCA is supported by scientific research, which has demonstrated its effectiveness in clinical trials. 42 Pruritus, a common adverse reaction influenced by the dosage administered, may result in the cessation of treatment and impact the optimization of the patient care. 43
B. Rifampicin
Rifampicin, a pregnane XR agonist, is the second-line therapy for CP patients who do not respond adequately or tolerate bile acid sequestrants. Sometimes, it is given to patients with ICP alongside UDCA. Rifampicin downregulates autotaxin (ATX) expression leading to reduced formation of lysophosphatidic acid (LPA), a potential pruritogen. 44
Studies on the efficacy of rifampin
Several studies have investigated the effectiveness of rifampin. One RCT evaluated the clinical efficacy of rifampin at a dose of 10 mg/kg/day in patients with PBC. The severity of pruritus was assessed using a predefined itch score: 3+, continuous, disturbing sleep pattern; 2+, moderate, not interfering with sleep pattern; 1+, mild intermittent, not affecting patient’s routine or sleep pattern; and 0, no itching. After 14 days of treatment, rifampin improved pruritus in all patients and was associated with significant decreases in liver biochemistries and s-BA levels (p < 0.05). Furthermore, after 3 months of treatment, itch disappearance was observed in 11 patients, and in all patients, the itch disappeared after 1 year of treatment. 45
A second RCT was conducted over 8 weeks on outpatients who received 14 days of treatment with rifampin 300–450 mg/day and placebo with a 14-day washout period between treatments. The severity of pruritus was assessed daily using VAS ranging from none (0) to severe (100), and the number of 4-g packs of cholestyramine ingested every 24 h. After the study, eight of nine patients who completed the clinical trial preferred rifampin to placebo (p = 0.03), and a significant reduction in pruritus was observed with rifampin compared to placebo (p < 0.002). The number of cholestyramine ingested did not change significantly. 46
A third RCT evaluated the anti-pruritic effects of rifampicin 10 mg/kg compared to phenobarbitone 3 mg/kg in PBC patients. The treatment was given for 14 days, with a 30-day washout period between treatments. Pruritus improved in 19 patients taking rifampicin but in only 8 patients taking phenobarbitone, and the itch improvement was substantially significant with rifampin than with phenobarbitone (p < 0.001). Pruritus disappeared in nine patients receiving rifampicin; of these nine patients, three patients were free of itch when they started phenobarbitone. Only rifampin reduced s-BA levels. The severity of pruritus at the start of treatment with phenobarbitone was also lower in patients who had crossed over from the rifampicin to the phenobarbitone group (p = 0.06). Prior treatment with phenobarbitone did not influence the degree of pruritus at the start of therapy with rifampin. 47
A fourth randomized study investigated the efficacy of rifampin 600 mg in patients with PBC. Pruritus was scored on a scale from 0 to 100. With rifampin, pruritus disappeared in 11 patients and partially improved in 3 patients; with placebo, only 2 patients had a partial response (p < 0.001). The study found six patients with a preceding poor or no response to cholestyramine improved with rifampin. In addition, rifampin, administered over 8 months, maintained relief from the pruritus. 48
A fifth RCT compared the efficacy of sertraline (100 mg/day) to rifampin (300 mg/day) in PBC and PSC patients with pruritus; both interventions relieved pruritus, but neither was superior in relieving pruritus (p-value = 0.740). 49
In another RCT in patients with non-A- and non-B hepatitis, alcoholic cirrhosis, PBC, or PSC, it was reported that rifampin 300 mg OD failed to relieve pruritus compared to a placebo. 50 Commonly reported side effects from rifampicin were hepatotoxicity, nephrotoxicity, hemolysis, and discoloration of body fluids.45,47,48
C. Fibrates activate the peroxisome proliferator-activated receptor-alpha and possess beneficial properties, such as anti-inflammatory, anti-cholestatic, and anti-fibrotic effects. These properties are believed to modify the progression of cholestatic liver disease, particularly PBC. They are currently used as an off-label treatment for PBC, usually as a complementary therapy with UDCA or bile acid sequestrants, and are generally considered an excellent alternative to rifampicin which can cause substantial drug–drug interactions by inducing various cytochrome P-450 (CYP) enzymes resulting in hepatotoxicity in up to 12% of patients if used for extended periods. 1
Studies on the efficacy of fibrates
A clinical trial using a randomized placebo-controlled design was conducted to investigate the effectiveness of bezafibrate (400 mg/day) in combination with continued treatment using UDCA. The main objective was to determine if there was a complete biochemical response, defined as normal levels of total bilirubin, alkaline phosphatase, aminotransferases, albumin, and prothrombin index after 24 months. The primary endpoint occurred in 31% of the patients assigned to bezafibrate plus UDCA compared to 0% in patients assigned to UDCA plus placebo [difference of −31% (95% CI: 10–50%; p < 0.001]. The study also found no significant difference in the mean itch intensity score between the bezafibrate and placebo groups from baseline to 24 months, with a mean difference of −95% (95% CI: −241 to 50%; p > 0.050). 51
In a non-randomized study, the efficacy of bezafibrate (400 mg/day) as an adjunctive treatment to UDCA (13–16 mg/kg/day) was evaluated in 48 PBC patients who exhibited an inadequate response to UDCA monotherapy. The study examined alterations in clinical characteristics, liver biochemistry, prognosis after treatment, and pruritus, which was assessed using the VAS (43 patients) and the 5-D descriptive pruritus scale. Following a median of 38 months, all but one of the patients with pruritus reported partial or complete relief of itching. After bezafibrate was discontinued, itching recurrence or aggravation was observed and five patients (23%) developed disease progression events. The combined therapy was also primarily effective in patients with less severe fibrosis and cholestasis severity. 52
Clinical trials investigating the use of fenofibrate in CP are currently unavailable. However, a retrospective study was carried out to examine the impact of fibrates (fenofibrate 200 mg/day or bezafibrate 400 mg/day) in combination with UDCA on PSC patients who did not respond adequately to UDCA monotherapy. Pruritus was documented using a scale ranging from 0 to 3, where 0 indicates the absence of pruritus, while 1, 2, and 3 represent mild, moderate, and severe pruritus, respectively. Fibrates combined with UDCA resulted in significant biochemical improvement. About 88% of the patients experienced significantly decreased pruritus intensity, including 3 who reported complete remission (p = 0.021). In one patient with complete remission, discontinuation of fibrates was followed by a recurrence of itching. 53
Alternative CP treatments when first- and second-line therapies have failed or are not feasible
Opioid receptor-mediated agents increase opioidergic tone in the central nervous system has been proposed to mediate CP. It has been theorized that opioid peptides cause pruritus by degranulating cutaneous mast cells or through a direct central and peripheral pruritogenic effect by activating MOR. 54 They are generally adjunct therapy in CP.
Studies on the efficacy of naltrexone
Naltrexone is the first choice among opioid receptor-mediated agents for CP treatment.
In one placebo-controlled study, patients were randomized to receive either 50 mg/day of naltrexone or a placebo for 2 weeks, followed by a 1-week washout. Then they crossed over to the other therapy for an additional 2 weeks. The study found that pruritus, as assessed daily using VASs, showed more significant improvements with naltrexone than with placebo (p < 0.0003). Forty-five percent of patients who received naltrexone experienced a decrease in pruritus of over 50% compared to baseline, and 5 experienced a complete disappearance of pruritus. 55
Another RCT also assessed the antipruritic effect of naltrexone at a dose of 50 mg/day. Pruritus scores, side effects, and liver function were evaluated every 2 weeks. The study found a significant reduction in mean changes in VASs from baseline for both daytime and nighttime itching in the naltrexone group (−54 versus −8%, p < 0.001 and −44 versus −7%, p = 0.003, respectively). 56
A third randomized study compared the efficacy of naltrexone with that of UDCA and ondansetron for managing acute cholestatic viral hepatitis and CP (severe pruritus defined as VASs >5). The primary endpoint was a reduction in VASs by at least 3 points from the baseline. All patients were re-assessed on day 5 of treatment, and a significant decrease was observed in all treatment arms (mean reduction in VASs was 4.5, 4.2, and 3.8, respectively, p < 0.009). However, the naltrexone group had a higher percentage of patients with significantly reduced itch intensity scores than the UDCA and ondansetron groups (88 versus 52% each). The administration of naltrexone was occasionally associated with a transient opioid withdrawal-like phenomenon that typically resolves spontaneously within 24–48 h. 57
Studies on the efficacy of naloxone
In an RCT that evaluated the efficacy of 1–2 continuous naloxone infusions (0.2 µg/kg/min) for CP, pruritus was assessed by four hourly recordings of VASs, and scratching activity independent of gross body movements was continuously recorded using a device that measured the frequencies associated with scratching activity. The study found that naloxone infusions were consistently associated with decreased scratching activity, ranging from 29 to 96% (mean: 50%; p < 0.001), and in 50% of these patients, the infusions were associated with a decrease in VASs. 58
Another RCT evaluated the effects of naloxone infusions in patients with CP. The study recorded pruritus scores (VASs maximum, 10.0) every 4 h while patients were awake, and scratching activity was recorded continuously. The study found that the mean VASs during naloxone infusions were 0.582 lower than that recorded during placebo infusions (95% CI: 0.176–0.988; p < 0.01). Additionally, the ratio of the geometric mean hourly scratching activity (HSA) during naloxone infusions to that during placebo infusions was 0.727 (95% CI: 0.612–0.842; p < 0.001). 59
In a prospective uncontrolled study, the role of intravenous naloxone in severe pruritus (measured by VASs 0−100) and associated symptoms of acute cholestasis were investigated. The study discovered that 81.8% of patients had a significant reduction in VASs after naloxone 0.4 mg was administered every 8 h for at least 48 h, and no adverse effects were observed. 60
Studies on the efficacy of nalfurafine
Nalfurafine is a drug that acts as an agonist of K-selective opioid receptors. It has been evaluated in several studies for its efficacy in treating pruritus in different conditions.
A post-marketing prospective study investigated the efficacy of nalfurafine in treating refractory CP. Records showed that 43 patients (97.7%) had previously undergone treatment with UDCA, while six patients (13.6%) had received bezafibrate. Participants received 2.5 μg nalfurafine OD for 12 weeks, and their pruritus scores were measured using the PBC-40 itch domain scores, VASs, and generic health-related quality of life (HRQoL). The study found that at week 12, the mean pruritus scores declined from baseline (PBC-40: 8.56–7.63; p = 0.041 and VASs: 42.9–29.3; p = 0.001), indicating the effectiveness of nalfurafine in alleviating refractory CP. 61
An interventional study on refractory pruritus evaluated nalfurafine hydrochloride for its clinical efficacy. Pruritus was assessed based on pruritus scores. Changes in VASs at week 4 were significantly greater in the nalfurafine hydrochloride 2.5 μg and 5 μg groups (28.56 and 27.46 mm, respectively, p = 0.002) compared to the placebo group (19.25 mm, p = 0.006). 62
Another clinic trial evaluated the effectiveness of nalfurafine hydrochloride in treating CP; the study used pruritus scores by Kawashima’s criteria and VASs to determine the severity of itching. Of the 24 refractory pruritus patients treated with nalfurafine, 17 (71%) indicated an improvement of itch, defined as a decrease in the VAS score ⩾30 mm. Also, all patients who received nalfurafine exhibited improved Kawashima scores ⩾1 point during the daytime or nighttime. 63
In a retrospective study that assessed the effectiveness of nalfurafine in treating CP, the study used pruritus scores by Kawashima’s criteria and VASs to determine the severity of itching. Results indicated that 67.4% of the participants administered nalfurafine achieved the primary endpoint, defined as a reduction in VAS of 50 mm or more. 64
Another interventional study evaluated the long-term efficacy and safety of nalfurafine in CLD patients with refractory pruritus. Patients were orally administered nalfurafine hydrochloride (2.5 μg/day) and repeatedly completed the same questionnaires and VASs for the entire follow-up period. Results showed that pruritus completely disappeared in approximately 39% (7/18) of patients, and VASs decreased over time in all patients who received nalfurafine. In addition, about 82% (9/11) patients followed up for >12 weeks showed continuous improvement of symptoms, and the progress was still apparent at ⩾20 weeks after starting nalfurafine (p < 0.0001). 65
Similarly, a preliminary prospective confirmatory trial evaluated the efficacy of nalfurafine in CP. Patients were pretreated with nalfurafine for ⩾4 weeks, and those who confirmed improvement in pruritus (decrease VAS of ⩾50 mm) were classified into two groups. The pruritus recurrence rate (increase in VASs of ⩾25 mm) was 100% in the discontinuous group and 0% in the continuous group, indicating that continuous treatment with nalfurafine is necessary to maintain the drug’s effectiveness in treating pruritus. 66 Adverse drug reactions were reported with nalfurafine, including pollakiuria, somnolence, insomnia, and constipation. 62
Studies on the efficacy of nalmefene
An opioid antagonist, nalmefene, is currently available for investigational purposes only.
In an RCT, oral nalmefene’s efficacy was evaluated for refractory CP treatment. The study measured scratching activity continuously for 24 h before and after each treatment period. Results showed that nalmefene therapy at doses of up to 20 mg BID was associated with a reduction in scratching activity by an average of 75% and a reduction in the mean VASs in all patients in the treatment group by an average of 77% (p < 0.01). 67
In an open-label trial, the efficacy of oral nalmefene therapy for refractory CP was further evaluated. Patients were given an initial ameliorating dose of nalmefene (2 mg BID), with incremental dosing of up to 30–120 mg BID. Scratching activity was recorded continuously for 24-h periods before and during treatment, while VASs were recorded every 4 h while patients were awake. Results showed that 13 out of 14 patients reported improved perception of pruritus with nalmefene therapy. In five patients, exacerbations of pruritus occurred approximately 4 weeks after an initial ameliorating dose had been reached, which was managed by increasing the nalmefene dosage. During nalmefene therapy, the mean VASs and scratching activity were lower in 13 and 12 patients, respectively, compared to the mean baseline values (p = 0.002 and p = 0.013, respectively). A transient opioid withdrawal-like reaction was observed with nalmefene therapy. 68
The fourth-line therapeutic option for patients with CLD who do not exhibit a response to or experience intolerance to rifampicin, fibrates, or opioids mediated agents.
Studies on the efficacy of sertraline
Sertraline is a selective serotonin reuptake inhibitor (SSRI) commonly used as an adjunct therapy for severe or refractory CP.
An RCT was conducted to determine the dose of sertraline and its effectiveness for CP in subjects with CP. The severity of pruritus was measured using VAS 0–10. Other factors such as distribution, timing, degree of disability, and physical evidence of scratching were also assessed. The study found that VASs improved with sertraline 75–100 mg/day but worsened in the placebo group (p = 0.009). Furthermore, changes in itch distribution, duration, direction, and physical evidence of scratching correlated with changes in the VASs. 69
Another interventional study evaluated the effectiveness and safety of sertraline as an adjunct therapy to treat pediatric refractory pruritus (ALGS and PFIC). The severity of pruritus was measured using VASs, skin scratch marks score, and a sleeping impairment score. Sertraline was prescribed with an initial dose of 1 mg/kg/day and increased gradually to 4 mg/kg/day. After 3 months, pruritus improved in 14 of 20 treated patients, and the median VASs decreased significantly from 8 (range 5–10) to 5 (range 2–10). Among the 14 patients with improved pruritus, 9 showed improvement in skin scratch marks and sleep quality scores. 70
Another RCT evaluated the long-term efficacy of sertraline on refractory pruritus in a large cohort with PBC. The severity of pruritus was assessed through self-reporting. The study showed that in 28 of 32 patients, the itching became stable or slightly changed over the follow-up period, while 4 patients experienced a sustained resolution of their pruritus. Additionally, among seven subjects who took sertraline for more than 6 months, 6 of 7 (86%) reported a significant reduction or resolution of pruritus in their weekly diaries, with a decreased or absent need for adjunct antipruritic medications. 71 A third RCT comparing sertraline to rifampicin found neither superior to the other. 49
Investigational therapies for patients with CLD who do not exhibit a response to standard CP treatments
Of 30−90% of patients who experience moderate to severe CP, 72 5−10% will develop pruritus refractory to conventional treatments; thus, investigational approaches may be necessary to relieve refractory pruritus. 44
Studies on the efficacy of ondansetron
A placebo-controlled trial evaluated the effectiveness of ondansetron (4 or 8 mg) in reducing VASs by at least 50% within 2 h of injection. The study found that ondansetron 8 mg effectively reduced VASs up to 6 h after injection, and the effect was consistent in the same patient. 73 A randomized cross-over study investigated the impact of ondansetron 8 mg TID on patients with CLD and CP. Patients were randomly assigned to receive either a placebo or ondansetron for 1 week, followed by a 1-week washout period, and then switched to another treatment for another week. The severity of pruritus was evaluated using CP-VASs. During the first week of therapy, ondansetron was found to decrease CP-VASs by 1.34 points (95% CI: 0.12–2.56; p = 0.033) in comparison to the placebo. A period effect was observed where the CP-VAS score decreased by 1.26 points (95% CI: 0.04–2.48; p = 0.044) during the second treatment period, regardless of the type of treatment used. Despite the improvement in itching observed with ondansetron treatment, patients did not prefer ondansetron over the placebo. 74
Another RCT examined the effectiveness of oral ondansetron in reducing pruritus associated with chronic liver disease. The study evaluated subjective pruritus scores and objective measurements of scratching activity over 24 h. The results showed that ondansetron therapy effectively reduced pruritus in 5 out of 13 patients (38%). The mean reduction in VASs was 27%, which was statistically significant (p < 0.05). However, the reduction in subjective itch intensity did not correlate to a substantial decrease in objective scratching activity (p = 0.19). 75 A fourth clinical trial that compared ondansetron (8 mg/day) to a placebo found no significant differences in mean pruritus scores or scratching activity. 76
Studies on the efficacy of the extracorporeal liver support system
A. Molecular Absorbent Recirculating System
The Molecular Absorbent Recirculating System (MARS) is an extracorporeal closed-loop albumin-dialysate circuit that incorporates the mechanisms of dialysis: ultrafiltration and adsorption to eliminate albumin-bound and water-soluble toxins from the blood. 77 An interventional study investigated the use of MARS for treating refractory CP in patients who had undergone liver transplantation. The study included nine patients, and the mean baseline VASs was 9.7 ± 0.5, which decreased to 3.7 ± 0.8 after treatment with MARS. In addition to the improvement in pruritus, the study found that patients who responded to therapy had a reduction in their mean baseline serum bilirubin levels (from 19.11 ± 16.96 mg/dL to 9.24 ± 3.52 mg/dL) and a reduction in the mean baseline s-BA levels (from 192.67 ± 58.12 μmol/L to 42.33 ± 31.58 μmol/L). During the follow-up period, three patients showed sustained improvement of pruritus lasting more than 3 months, while pruritus relapsed in three patients. 5
Another experimental study evaluated MARS in PBC patients with resistant pruritus. The severity of pruritus, transaminases, and s-BA levels was measured before and after treatment and 30 days after treatment. The results showed a significant decrease in pruritus severity in all but one patient [mean VASs: 20.1 ± −4.2, 70.2 ± 4.8 (baseline), p < 0.001], with a reduction of 72% immediately after treatment and 51% after 1 month. Additionally, a significant decrease in s-BA levels was observed after treatment and after 1 month. The effect of MARS on pruritus and cholestasis markers was similar in patients irrespective of the etiology of CP. 78
In another intervention study, the effects of MARS on patients with acute liver failure (LF), acute-on-chronic LF, and intractable CP were assessed over an 8-year follow-up period . The study found significant improvements in serum bilirubin, transaminases, and encephalopathy grade. Among the 38 patients with acute LF who were listed for liver transplantation and received MARS, 20 died on the waiting list, and 18 survived. Of the 18 who survived, 14 recovered on MARS therapy without transplantation. 79
In a prospective study of three HCV-cirrhotic patients with intractable pruritus who had failed medical treatment, MARS therapy resulted in subjective improvement in pruritus and quality of life and decreased s-BA levels. The patients did not require retreatment or liver transplantation up to a 9-month follow-up. 80
In a fifth study, MARS therapy was evaluated in PBC patients with intractable pruritus. Pruritus, scratching lesions, and standard liver tests, including s-BA levels, were measured. The study found that MARS therapy was associated with substantial relief of itching and scratching, with pruritus quickly resolving in two patients and markedly improving in the other two. The s-BA levels decreased in all patients, and no patient needed retreatment up to a 7-month follow-up. 81
A retrospective study on the efficacy of MARS in severe CP was conducted among three children who received a total of 135 MARS sessions before liver transplantation. Total bilirubin, s-BA levels, and pruritus (NRS 0 = no pruritus, 10 = maximal pruritus) were assessed. Pre-MARS s-BA concentrations averaged 207 ± 67 μmol/L. After MARS therapy, a significant reduction in s-BA levels from baseline to 67 ± 9%, 48 ± 3%, 38 ± 14%, and 37 ± 5%, respectively, within 2, 4, 6, and 8 h of therapy (p < 0.05) and a reduction in the mean itch-NRS score decreased [3.3 ± 2.9 versus 6.5 ± 2.3 (baseline), p < 0.01]. 82
MARS was reported by another study as a safe therapeutic option for significantly relieving refractory CP in the first or second treatment. 83
However, a study involving nine hepatitis C patients with intractable pruritus treated with MARS did not find a statistically significant decrease in total bilirubin, creatinine, s-BA levels, and VASs. 84 No significant adverse effect was observed with MARS therapy.80–82
B. Charcoal hemoperfusion
An extracorporeal technique like MARS, was also investigated by a study. The retrospective study involving nine patients showed that charcoal hemoperfusion significantly decreased pruritus in 69% of patients. The median NRS rating decreased from 9/10 to 4/10 (p = 0.004), indicating a significant reduction in pruritus severity. Six patients returned for follow-up, and symptom-free periods ranged from 8 to 90 days, with a median of 18 days. Common adverse reactions were pain, bleeding from the catheter site, and fever. 85
Fractionated plasma separation and absorption, also called Prometheus, is an extracorporeal liver support system with direct toxin adsorption of the patient’s albumin fraction. Prometheus has recently been introduced; hence studies on Prometheus are limited.
An interventional study included seven patients. Their pruritus severity was evaluated before and after Prometheus treatment using VAS ranging from 0 (no pruritus) to 10 (severe pruritus) and by measuring s-BA and total bilirubin levels. After treatment with Prometheus, pruritus severity was significantly decreased, with VASs dropping from 9 ± 1 to 3 ± 3 (p < 0.001). Additionally, s-BA levels decreased from 248 ± 192 µmol/L to 101 ± 85 µmol/L (p < 0.03); 4 weeks after treatment, four patients still experienced a distinctive benefit. In contrast, two patients’ itch relief lasted only a few days, suggesting that Prometheus therapy can effectively reduce pruritus severity in patients with various liver diseases. Still, its long-term effects may vary. 86
C. Plasmapheresis
An experimental study involved three PBC patients with intractable pruritus who underwent plasma separation and anion adsorption for three consecutive days. The primary outcome measures were pruritus severity [defined as pruritus ⩾7 on a rating scale (0 (no pruritus) to 10 (maximal pruritus)) on at least 4 out of 7 days despite medical treatment], fatigue was assessed using the Fisk Fatigue Severity Score, and the PBC-40, a disease-specific HRQoL measure. Plasmapheresis resulted in a transient improvement in pruritus severity, with a mean score reduction of 46% and 24% at weeks 1 and 4, respectively. However, there was no significant improvement in fatigue or QoL, and s-BA levels did not correlate with pruritus intensity. 87
Another experimental study evaluated the role of plasmapheresis in five PBC patients with refractory pruritus, hypercholesterolemia, and xanthomatous neuropathy. Plasmapheresis caused a marked improvement in pruritus (mean decrease in VAS ~80%) and reduced fatigue. Cholesterol levels were lowered at an average of 10.3 mmol/L, xanthomata, and the painful xanthomatous neuropathy was decreased in three of four patients. Repeat sessions of plasmapheresis sustained its effect. 88
A third interventional study that evaluated the antipruritic effect of plasmapheresis in refractory CP observed a mean reduction [3.1 ± 2.2 versus 8.3 ± 1.4 (baseline), p < 0.0001] in the 10-point itch-NRS score after treatment. In addition, there were significant decreases in serum transaminases and bilirubin levels, with the antipruritic effect persisting throughout the 90-day follow-up. 89
D. Biliary drainage via ileal exclusion (IE), partial internal or external biliary diversion (PBED), endoscopic nasobiliary drainage (ENBD), and endoscopic stenting (ES) are invasive procedures that have been investigated for the treatment of refractory pruritus in children with CLD. An interventional study investigated PBED as the primary procedure to treat children with intractable pruritus and chronic intrahepatic cholestasis. At 3–8 years follow-up, patients with PFIC were found to be free from itch since surgery, and s-BA concentrations fell significantly after surgery (218−275 µmol to <10 µmol). Additionally, biochemical tests of liver function and histology returned to normal or near normal, and the quality of life after surgery was excellent. 90
A retrospective study evaluated the efficacy of IE in children with refractory pruritus and chronic intrahepatic cholestasis. The authors found that 72.7% of patients experienced complete resolution or significant reduction of pruritus, allowing for improved biochemical parameters and survival with the native liver. 91
Another study also investigated IE as an alternative therapeutic option to PEBD for children with PFIC. In group 1, comprising four children, IE was the primary procedure, and in five children, IE was performed after PBED (group 2). In the first group, three children experienced alleviation in pruritus with decreased bilirubin and s-BA concentrations 2 and 5 years after IE, while one had to be converted to PEBD. After 10 years, only 2 children remained available for further examination. Both children exhibited varied degrees of itching and showed elevated s-BA levels. In group 2, 1 ultimately required transplantation and another developed varying degrees of pruritus, while 3 others had excellent outcomes; suggesting IE is an alternative rescue option for patients who cannot benefit from PEBD. 92
In a retrospective study, the outcome of PIBD for intractable pruritus in children with CLDs (PFIC and AGLS) was evaluated. The study found that pruritus resolved in 9 children with a significant reduction of s-BA levels (p < 0.02). 93
In another retrospective study, the safety and efficacy of ENBD for refractory CP were assessed. The authors found that ENBD decreased pruritus in 89.6% of patients [mean VASs 0.3 versus 10.0 (baseline), p < 0.0001] with about 33% of patients free of pruritus within 24 h of initiating NBD. Significant improvements were also seen in the serum levels of alkaline phosphatase (p = 0.001) and serum bilirubin (p = 0.03). 94
Similarly, an interventional study examined the safety and effectiveness of short-term ES for symptomatic dominant strictures in PSC patients. Primary endpoints were changes in complaints and cholestasis after 2 months and the time interval until a repeat endoscopic treatment was deemed necessary. Cholestatic complaints improved after 2 months in 83% of patients, and the mean levels of conjugated bilirubin, alkaline phosphatase, and gamma-glutamyl transpeptidase dropped significantly (p < 0.05). Additionally, the reintervention-free proportions were 80% and 60% in the first- and third-year post-ES. 95
The biliary drainage procedures were relatively safe, with transient endoscopic retrograde cholangiopancreatography pancreatitis being the most common complication.90–95
Studies on the efficacy of phototherapy
Two studies evaluated different types of light therapy for refractory CP. The first study, an open-label pilot study on bright-light treatment, was administered twice daily for approximately 60 min at a dosage of 10,000 lux. The study included eight patients with diverse etiologies of CP. The pruritus intensity was measured using VASs and a monitoring system that recorded HSA. In seven out of eight patients, a non-statistically significant reduction in the mean HSA (−32.2%; p = 0.123), and mean VASs were lower in six patients (−42%; p = 0.05) was noted. 96
The second study, was an observational study on ultraviolet B (UVB) phototherapy for CP. The study found that 77% of patients experienced a >60% reduction in perceived pruritus. VASs decreased significantly from 8.0 to 2.0 (p < 0.001). In addition, in four patients who required an extra course of phototherapy due to recurrent CP, marked improvement in pruritus was observed after the repeat session. UVB did not significantly change cholestatic serum markers. 97
Studies on the efficacy of the ileal bile acid transporter inhibitor
IBAT inhibitors are currently being investigated for their potential use in managing refractory CP. They interrupt the enterohepatic cycle of bile acids, thereby markedly reducing the reuptake of bile salts from the small intestine−terminal ileum. This can benefit patients with certain liver and bile-related disorders, including PFIC and ALGS, characterized by inherited defects in bile acid transporters. Currently, two FDA-approved IBAT inhibitors are available on the market: odevixibat, used to treat CLDs (including PFIC9), and maralixibat (MRX), used to treat chronic refractory CP in ALGS.
Odevixibat (A4250)
Two phases 3 trials (PEDFIC 1 and PEDFIC 2) investigated the efficacy of odevixibat in PFIC patients with pruritus. In PEDFIC 1 study, the effectiveness and safety of odevixibat (A4250) at 40 and 120 µg/kg OD were compared to a placebo in children with PFIC types 1 and 2. The primary endpoint was the proportion of positive pruritus assessments (PPA), which was defined as a scratching score of ⩽1 or ⩾1 point drop from baseline or the percentage of participants experiencing at least a 70% reduction in baseline fasting s-BA concentration or reaching a level ⩽70 μmol/L after 24 weeks of ileal bile acid transporter inhibitor (IBATi). The severity of observed scratching in the AM and PM was scored by Albireo Observer-reported Outcome (ObsRO) instrument (0−4 where 0 is no scratching and 4 is the worst possible itch). In this study, the mean PPA was significantly higher with odevixibat versus placebo [58% in the 40 μg/kg/day group versus 52% in the 120 μg/kg/day group versus 30% in the placebo group with a mean difference of 25·0% (95% CI: 8.5–41.5; p = 0·0038)]. More patients achieved a fasting s-BA response with A4250 40 µg/kg/day [14 of 42 patients (33%) in the combined odevixibat group (10 in the 40 μg/kg/day group and four in the 120 μg/kg/day group)] versus none of 20 in the placebo group [proportion difference 30·7% (95% CI: 12.6–48·8; p = 0·003)]. Odevixibat was generally well tolerated. Common side effects were diarrhea and vomiting at low and high doses.98,99
To evaluate the long-term safety and efficacy of odevixibat, a PEDFIC 2 trial, a 72-week extension study of PEDFIC 1 participants plus newly enrolled PFIC patients of any age administered A4250 at 120 µg/kg/day, is currently underway with data being collected on primary outcome measures such as a change in pruritus as indexed by caregiver-reported observed scratching on the Albireo ObsRO scale and s-BA levels.100,101
Similarly, a pilot study assessed the efficacy and safety of the A4250 in PBC patients undergoing bile acid sequestrant treatment. After a 2-week washout, they were treated with either 0.75 or 1.5 mg of A4250 for 4 weeks. VASs, 5-D itch scale, and PBC−40 questionnaires assessed patients’ pruritus severity and s-BA levels measured. All nine patients exposed to A4250 reported a remarkable improvement in pruritus according to 5-D itch, VAS, and PBC−40 pruritus, a promising result for odevixibat use in PBC which is a common CP disorder. 102
Maralixibat
The ICONIC trial was a phase 2b study with an open-label extension and a randomized withdrawal period (RWD) involving children aged 1–18 with ALGS. Participants were administered MRX at a daily dose of 380 μg/kg for 18 weeks. Afterward, they were randomly assigned to receive either a placebo or continue with MRX for 4 weeks. Subsequently, all participants received open-label MRX until week 48, during which the dose was increased to 380 μg/kg BID. The primary endpoint was the mean s-BA change during the RWD in participants, with a ⩾50% reduction in s-BA by week 18. CP was assessed using observer-, patient-, and clinician-rated scales ranging from 0 to 4. The results showed that participants who switched to placebo during the RWD experienced significant increases in s-BA levels (94 μmol/L, 95% CI: 23–164 μmol/L) and pruritus intensity (1.7 points, 95% CI 1.2–2.2 points). In comparison, those who continued MRX demonstrated a significant reduction in s-BA levels (−96 μmol/L, 95% CI: −162 to −31 μmol/L) and improved pruritus (−1.6 points, 95% CI: −2.1 to −1.1 points), with sustained effect during the long-term extension period of 204 weeks. 103
The ITCH trial, which involved children with ALGS, was a phase 2 study that was conducted in a double-blind manner. During the dose escalation phase of the study, participants were administered MRX in increasing amounts, starting with 14 μg/kg/week in the first week and gradually reaching a stable dose of either 70, 140, or 280 µg/kg/day for up to 5 weeks. This dosage was then continued up to week 13. The study’s primary objective was to compare the change in pruritus between the baseline and the end of week 13, relative to the placebo group. The primary outcome was the change in pruritus from baseline to week 13 relative to placebo. Pruritus was assessed by the caregiver [itch-reported outcome instrument (ItchRO)] and clinician report [range, 0–4 (severe)]. In this study, significant decreases were observed with doses of 70 µg/kg/day (mean adjusted difference, −0.89; 95% CI: −1.70 to −0.08; p = 0.032) and 140 µg/kg/day (mean adjusted difference, −0.91; 95% CI: −1.62 to −0.19; p = 0.014) but not with 280 µg/kg/day (mean adjusted difference, −0.04; 95% CI: −0.94 to 0.86; p = 0.44). A 1-point reduction in pruritus occurred more commonly with MRX versus placebo participants (caregiver ItchRO, 65% versus 25%; p = 0.06; clinician score, 76% versus 25%; p = 0.01). There were no significant changes in liver chemistries or s-BA levels relative to the placebo. 104
INDIGO, an open-label phase 2 study of MRX’s efficacy and long-term safety (up to 280 µg/kg/day OD or BID) in treating CLD in 33 pediatric patients with PFIC (aged 12 months to 18 years), is ongoing. A 4-week dose escalation period was followed by a 4-week stable dosing period, then a 5-week stable dosing period, continued by a 59-week long-term exposure period, and an optional follow-up treatment period for eligible participants who continued treatment with MRX. A 48-week interim data analysis showed the mean serum s-BA reduction at weeks 13 and 48 was 29 and 59 μmol/L, respectively. Six patients experienced s-BA normalization (⩽8.5 µmol/L) or reduction from baseline by ⩾70% and ItchRO of zero or substantial improvement ⩾1.0 point (responders). Treatment-emergent adverse events were reported in all patients. The most frequent side effects were pyrexia, diarrhea, cough, and abdominal pain. 105
The randomized, double-blind, placebo-controlled phase 2 IMAGO, IMAGINE, and IMAGINE-II long-term extension studies also showed that a proportion of patients with ALGS treated with MRX experienced improvements in their s-BA levels and pruritus.106–108
A phase 2 study was conducted to evaluate the efficacy of MRX in treating CP related to PBC. Participants taking UDCA for at least 6 months or who could not tolerate it were randomly assigned to either MRX (10 or 20 mg/day) or a placebo for 13 weeks, in conjunction with UDCA if possible. The study’s primary endpoint was the change in the average weekly sum score of ItchRO from baseline to week 13 or early termination (ET). The mean ItchRO weekly sum scores for MRX and placebo decreased from baseline to week 13/ET, with a difference between the two groups that was not statistically significant (p = 0.48) (−26.5; 95% CI: −31.8, −21.2 and −23.4; 95% CI: −30.3 to −16.4, respectively). 109
MARCH−PFIC, a double-blind placebo-controlled phase 3 trial, is ongoing to evaluate the efficacy and safety of MRX (up to 600 µg/kg BID) in subjects with PFIC (aged >12 months and <18 years). The primary outcome measure is the treatment response as measured by the mean change in pruritus severity as assessed by ItchRO [Obs] between baseline and weeks 15 through 26. Running concurrently is MARCH-ON, an open-label extension study to evaluate the long-term safety and efficacy of MRX (up to 600 µg/kg BID) in treating subjects with PFIC aged 1–18 years who completed MARCH−PFIC to determine the incidence of treatment-emergent adverse events from baseline through study completion (up to 104 weeks). 110
MRX-800 an open-label phase 2 trial in subjects who have previously participated in the MERGE study evaluating MRX in the treatment of CLD, including, but not limited to, ALGS, PFIC, and biliary atresia is also ongoing. 111 Another open-label phase 2 RISE trial is underway to assess the safety and tolerability of MRX in infants <12 months of age with ALGS or PFIC. The EMBARK Phase 2b study is ongoing to evaluate MRX efficacy and safety in infants with biliary atresia after hepatoportoenterostomy. 112
However, the estimated yearly cost of IBATi, for instance, MRX for a 17 kg patient, is over $396,000 ($113,150−$1,697,250), 26 alongside the reported adverse effect of dose-dependent diarrhea and abdominal discomfort could potentially impede long-term use in treating CP. 113
Linerixibat (GSK2330672)
Several randomized, placebo-controlled clinical trials have also begun to determine the efficacy of IBATi in common CLDs – for instance, the GLIMMER/VANTAGE trial in PBC patients with moderate and severe pruritus. GLIMMER phase 2a study evaluated the efficacy of short-term administration of linerixibat (GSK2330672) for treating PBC patients with pruritus. The primary endpoints were the safety of GSK2330672, assessed using clinical and laboratory parameters, and tolerability as rated by the Gastrointestinal Symptom Rating Scale. The secondary endpoints were changes in pruritus scores measured using the 0–10 NRS, primary biliary cholangitis-40 (PBC-40) itch domain score and 5-D itch scale, changes in serum total bile acids, etc. GSK2330672 treatment for 14 days was safe, with no serious adverse events reported. After GSK2330672 therapy, the percentage changes from baseline itch scores were −57% (95% CI: −73 to −42, p < 0·0001) in the NRS, −31% (−42 to −20, p < 0·0001) in the PBC-40 itch domain, and −35% (−45 to −25, p < 0·0001) in the 5-D itch scale. GSK2330672 produced a significantly more significant reduction from baseline than the double-blind placebo in the NRS (−23%, 95% CI: −45 to −1; p = 0·037), PBC-40 itch domain, (−14%, −26 to −1; p = 0·034), and 5-D itch scale (−20%, −34 to −7; p = 0·0045). After GSK2330672 treatment, s-BA levels declined by 50% (95% CI −37 to −61, p < 0·0001) from 30 to 15 μM. 114
GLIMMER phase 2b dose-response study evaluated the long-term efficacy, safety, and tolerability of linerixibat (GSK2330672) for treating PBC patients with pruritus. One hundred forty-seven patients received linerixibat doses – 40 mg BID, 90 mg BID, 180 mg OD, 20 mg OD, and 90 mg OD – or placebo. Treatment was a 4-week single-blind placebo period; patients with NRS ⩾3 were randomized to receive a placebo or linerixibat (double-blind) for 12 weeks, a 4-week single-blind placebo period, followed by a 4-week follow-up period. The primary objective was to investigate dose-related changes in the mean worst daily itch (MWDI) score from baseline at week 16 (0−10 NRS). Patients were randomized to receive a placebo or linerixibat (20 mg OD, 90 mg OD; 180 mg OD: 40 mg BID, 90 mg BID). Linerixibat groups showed ⩾2-point mean reductions in MWDI from baseline at week 16; however, the mean differences from placebo were insignificant. Post hoc analysis of change from baseline in monthly itch score over the treatment period showed significant differences between placebo and linerixibat 180 mg once daily (p = 0.0424), 40 mg twice daily (p = 0 .0105), and 90 mg BID (p = 0.0370). 115 Linerixibat 40 mg BID is being studied in the ongoing confirmatory Phase 3 GLISTEN trial. 116
Volixibat
Volixibat is currently under evaluation by two current studies. OHANA Phase 2a/b study evaluates the efficacy and safety of volixibat 20 mg and 80 mg BID in ICP with elevated s-BA concentrations. 117 On the other hand, the VISTAS Phase 2b trial evaluates the efficacy of volixibat 20 mg and 80 mg BID in treating PSC patients with pruritus and assesses the potential impact on the disease progression of PSC. 118
Miscellaneous investigational therapeutic options
Limited clinical trials have also observed some itch improvement with phenobarbital, corticosteroids, 119 colchicine, 120 methotrexate,120,121 flumecinol, 122 gabapentin, 123 lidocaine, 124 and propofol. 125
Liver transplantation
When conservative and experimental therapeutic options fail, liver (re)transplantation can be an effective treatment for patients with progressive liver dysfunction and severe portal hypertension 126 or for those whose liver function is preserved but develop pruritus that substantially impacts HRQoL or causes suicidal ideation. 10 In pediatric CLDs, the most common indication for liver transplantation is biliary atresia, accounting for 42–54% of pediatric liver transplants. 127 Nearly 22% of BA patients reach adulthood with their native liver, with half requiring liver transplantation later in life. 128 In adults, CLD is responsible for about 5.7–11% of all liver transplants, with PSC and PBC being the most common indications.129,130 Postoperatively, PBC and PSC have the most favorable outcomes, with 10-year survival rates of 79% and 83%, respectively, compared to other indications for liver transplantation. 131 However, autoimmune diseases, including PBC and PSC, notably recur in many liver transplant recipients and may be more aggressive than the original disease. 132 The recurrence rates of PBC range from 8 to 16% at 1–6 years, 21 to 37% at 10 years, and >43% at 15 years, while autoimmune hepatitis recurs in 25% at 5 years and 50% at 10 years. The recurrence rate of PSC ranges from 8.6 to 25%. The underlying mechanism of these recurrences remains to be determined, making ideal management challenging. 133
Strengths and limitations: The primary advantage of our research lies in its extensive analysis of current literature to derive broad theoretical conclusions on the efficacy of traditional and new therapies for CP. However, our research is not without limitations. Our analysis included studies that utilized various scales and tools to quantify pruritus, a complex construct which pose a challenge to making a direct comparison between studies that use different evaluation instruments to assess the efficacy of a particular drug, thus impeding the ability to draw reliable conclusions about treatment effectiveness and/or hindering the development of consistent treatment guidelines. In addition, our systematic review may be at risk of publication bias, where studies with significant findings are more likely to be published than those with negative or inconclusive results, thus overestimating the true effect of an intervention.
Conclusions
While no causal link has been established between CP and pruritogen accumulation in the plasma,134–136 several approaches starting with UDCA/bile acid binding resins, escalating to rifampicin or fibrates, SSRIs, and opioid antagonists, appear to be rational treatment options. While for refractory pruritus, more invasive solutions may be required. Recent clinical trials identify IBATi as a promising treatment option for CP, with phase 2–3 clinical trials demonstrating a durable improvement of itch in inherited childhood cholestatic disorders (PFIC and ALGS). Clinical trials also show promising potential for IBATi in alleviating pruritus associated with other prevalent CLDs, which can make them a viable alternative for refractory CP as a second-line treatment. As the efficacy of IBATi in the broader CP population awaits confirmation from ongoing randomized trials, the high cost of these drugs could limit their widespread usage.
Supplemental Material
sj-docx-1-tag-10.1177_17562848231172829 – Supplemental material for Systematic review: efficacy of therapies for cholestatic pruritus
Supplemental material, sj-docx-1-tag-10.1177_17562848231172829 for Systematic review: efficacy of therapies for cholestatic pruritus by Ebehiwele Ebhohon and Raymond T. Chung in Therapeutic Advances in Gastroenterology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.