
Abstract
Background:
Vedolizumab, a humanized antibody targeting the α4β7 integrin, was proven to be effective in the treatment of moderate-to-severe ulcerative colitis (UC) in randomized clinical trials. The aim of the POLONEZ study is to determine the demographic and clinical characteristics of the patients with UC treated with vedolizumab within the scope of the National Drug Program in Poland and to assess the real-world effectiveness and safety of vedolizumab in the study population. Here we report the demographic and clinical characteristics of these patients.
Methods:
This prospective study included adult patients eligible for UC treatment with vedolizumab who were recruited from 12 centers in Poland between February and November 2019. Collected data included sex, age, disease duration, presence of extraintestinal manifestations or comorbidities, status of previous biologic treatment, and current concomitant treatment. Disease extent was determined according to the Montreal classification, and disease activity was measured with the Mayo Score.
Results:
A total of 100 (55 biologic-naïve and 45 biologic-exposed) patients were enrolled in the study (51% female, median age 35 years). Among biologic-exposed patients (mostly infliximab-treated), 57% had failed to respond to the therapy. The disease duration was significantly shorter in biologic-naïve (median 5 years) than in biologic-exposed (8 years, p = 0.004) or biofailure patients (7 years, p = 0.04). In the overall population the median Total Mayo Score was 10. Disease extent and activity were similar between the subgroups.
Conclusions:
Our study indicates that patients treated with vedolizumab in Poland receive the drug relatively early after UC diagnosis, but their disease is advanced. More than half of the patients had not been treated with biologic drugs before initiating vedolizumab.
The study was registered in ENCePP database (EUPAS34119).
Lay summary
Treatment of moderate-to-severe ulcerative colitis (UC) with the integrin antagonist vedolizumab became available within the Polish National Drug Program (NDP) in 2018. In this study, for the first time, we provide detailed demographic and clinical characteristics of 100 patients (median age 35 years, 51% female) treated with vedolizumab in Poland, of whom 55 were biologic-naïve and 45 biologic-exposed. The median duration of disease was 6 years. The disease duration was shorter in biologic-naïve than in biologic-exposed patients. Most patients were affected by extensive colitis (52%) or left-sided colitis (42%). Median disease activity was 10 according to the Total Mayo Score. Sixty-eight patients received concomitant systemic corticosteroids and 45 patients received immunomodulators. Our findings indicate that Polish patients receiving vedolizumab have a high disease activity and are treated relatively early after UC diagnosis. This might be due to the criteria for inclusion of a patient in the NDP.
Introduction
Ulcerative colitis (UC) is a chronic inflammatory disease of the colon. Typical symptoms include bloody diarrhea, fatigue, and abdominal discomfort. 1 In Europe, the prevalence and incidence rates of UC are among the highest in the world, accounting for approximately 1.3–2.1 million people diagnosed with UC.2,3 The incidence of UC varies across European countries (between 0.9 and 24.3 cases per 100,000 person-years), with higher rates observed in northern and western countries than in eastern regions of the continent.3,4 Recently, however, growing rates of UC prevalence and incidence were observed in Central and Eastern European countries. 5 In Poland, data on the epidemiology of UC in the general population are not available, but increasing hospitalization rates associated with UC in recent years may indicate an increasing incidence rate of the disease. 6 The natural course of the disease is characterized by fluctuating periods of relapse and remission. 2 Conventional treatment of UC with 5-aminosalicylic acid (5-ASA) derivatives, oral immunomodulators, and corticosteroids may be ineffective and/or associated with unacceptable adverse events. 7
Antibody-based drugs targeting tumor necrosis factor alpha (anti-TNF), such as infliximab, adalimumab, and golimumab, considerably improved the management of UC. 8 Although anti-TNF drugs are effective at inducing and maintaining disease remission,9–12 up to 30% of patients do not respond to induction therapy, and up to 45% lose response during treatment. 13 In addition, the immunosuppressive action of anti-TNF drugs can be associated with serious adverse effects. 14 Therefore, new treatment options representing different mechanisms of action are needed.
Vedolizumab is a humanized monoclonal antibody that targets the α4β7 integrin, inhibiting inflammation in the intestinal mucosa. 15 To date, vedolizumab is the only monoclonal antibody registered for the treatment of UC that acts selectively in the gastrointestinal tract, in contrast to anti-TNF drugs, which exert systemic effects, and the α4 integrin antagonist natalizumab, which reduces inflammation by acting on α4β7 and α4β1 integrins. 15 The phase 3 GEMINI 1 study provided evidence for the efficacy of vedolizumab (with a response rate of 47.1% for induction therapy and a clinical remission rate of 41.8% for maintenance therapy with vedolizumab every 8 weeks)16 and led to its market authorization for the treatment of moderate-to-severe UC.
Real world evidence (RWE) studies are crucial to understanding the clinical characteristics of treated patient populations at large as well as the effectiveness and safety of treatments in daily clinical practice. To date, several real-world studies (most of them retrospective) addressing the effectiveness and safety of vedolizumab have been conducted, predominantly in the USA and Europe. 17 Their findings are consistent with those observed in clinical trials. 17 However, detailed information on the clinical profile of patients with UC who are offered biologic therapies is scarce, especially for Eastern European countries. In Poland, the eligibility of UC patients for a reimbursed biologic treatment, which is limited to infliximab and vedolizumab, is governed by the criteria set by the National Drug Program (NDP). 18 These criteria are stricter than the indications listed in the vedolizumab product characteristics officially approved by the European Medicine Agency (EMA). 19 This may affect the characteristics of the population of patients treated with vedolizumab in Poland, and, as a consequence, could possibly impact the outcomes of the therapy. Therefore, based on the data from the nationwide non-interventional, prospective POLONEZ study, we analyzed the baseline demographic and clinical characteristics of patients treated with vedolizumab in the setting of the NDP to get more insights and a better understanding of this patient population.
Patients and methods
Patients and study design
The multicenter, non-interventional, prospective POLONEZ study aims to determine the demographic and clinical characteristics of the patients with UC treated with vedolizumab within the scope of the NDP in Poland and to assess the effectiveness and safety of a 54-week therapy in the study population. The assessments are scheduled at weeks 14 and 54 of therapy, with a follow-up visit at week 80. Adult patients who were eligible for UC treatment with vedolizumab according to the local Summary of Product Characteristics19 and met the inclusion criteria of the NDP (fully reimbursed by the Ministry of Health in Poland), that is, had severely active UC, contraindications to treatment with ciclosporin, and an inadequate response to or intolerance or other contraindications to conventional therapy (including both corticosteroids and immunosuppressive drugs), were included in the study.18
Exclusion criteria, according to the local Summary of Product Characteristics and the NDP regulations, were as follows: hyperreactivity to vedolizumab or excipients; severe active or opportunistic infections (e.g. progressive multifocal leukoencephalopathy); chronic heart, kidney, liver, or respiratory failure; demyelinating disease; precancerous condition or malignancy diagnosed within 5 years prior to study enrollment; pregnancy; or breastfeeding.
Consecutive patients who were qualified for vedolizumab treatment within the scope of the NDP in each of 12 centers in Poland were enrolled in the study between February and November 2019. Baseline data collected included sex, age, disease duration, smoking status, UC-related hospitalizations within the past 12 months, presence and type of extraintestinal manifestations, comorbidities, previous UC therapy with biologic medications, and concomitant medications (i.e. corticosteroids, immunomodulators, 5-ASA derivatives). The extent of disease was determined according to the Montreal classification,20 and disease activity was measured with the Total Mayo Score (range 0–12, with higher scores indicating a more active disease). 21 In selected analyses, a Partial Mayo Score (Total Mayo Score without the endoscopic component; range 0–9) was applied. 22 Patients who did not improve after 4 weeks of corticosteroid treatment with a daily dose of up to 0.75 mg/kg body weight of prednisolone (or equivalent) were considered corticosteroid-refractory. 18 Corticosteroid-dependency was defined as the impossibility to reduce the daily corticosteroid dose below 10 mg of prednisolone equivalent within 3 months after corticosteroid initiation or disease relapse within 3 months after corticosteroid discontinuation.18
The study protocol was approved by the Bioethics Committee of the Maria Sklodowska-Curie National Cancer Institute (Approval No 79/2018). All patients gave written informed consent to participate in the study. The study was registered in the European Network of Centers for Pharmacoepidemiology and Pharmacovigilance (ENCePP) clinical trial database (EUPAS34119).
Statistical analysis
All statistical analyses were performed with R version 3.5 (R Foundation for Statistical Computing). 23 Continuous variables are shown as median and interquartile range (IQR) or range. For categorical variables, the number of observations and percentages are given. Groups were compared using the U Mann–Whitney test for quantitative variables and the chi-square test (Fisher’s test) for qualitative variables, with the significance level set to 0.05.
Results
Demographic characteristics and previous biologic treatment
A total of 100 patients were enrolled in the study. Both sexes were almost equally represented, and the median age of the enrolled patients was 35 years (range 18–82) (Table 1). Most patients never smoked, one in four quit smoking, and only 4 (4%) individuals in the total study group declared themselves as current smokers.
Demographic characteristics of the study population.

BMI, body mass index; IQR, interquartile range.
biologic-naïve vs biologic-exposed.
biologic-naïve vs biofailures.
U Mann–Whitney test.
chi-square test.
In the study population, 55 (55%) patients had not been treated with biologic drugs (biologic-naïve) and 45 (45%) patients received at least one dose of a biologic for UC prior to study enrollment (biologic-exposed). No statistically significant differences were observed in the demographic characteristics between biologic-naïve and biologic-exposed/biofailure patients (Table 1). Most biologic-exposed patients received anti-TNF treatment: 89% were treated with infliximab only, 4% with adalimumab only, and 4% with infliximab and adalimumab. In addition, one patient was treated with golimumab and vedolizumab within clinical trials.
All patients treated previously with infliximab and/or adalimumab had completed one course of induction treatment, and approximately one-third of patients treated with infliximab and one-half of patients treated with adalimumab underwent one course of maintenance treatment (Table 2, Figure 1). Among 44 patients previously treated with infliximab and/or adalimumab, the treatment had failed in 25 (57%) individuals (biofailures). The most common reason for early termination was primary lack of response to treatment. The other reason for early termination was intolerance to the treatment. Approximately one-third of patients treated with infliximab and two out of four treated with adalimumab achieved clinical remission. Of 32 patients treated with infliximab who had an endoscopic assessment, mucosal healing was observed in 12 (37.5%) individuals. No attempts at dose escalation were made.
Previous treatment of ulcerative colitis with biologic drugs (data collected retrospectively).

data for treatment with infliximab.
data for treatment with adalimumab.
endoscopic assessment was done in 31 patients on infliximab only, 1 patient on adalimumab only, and 1 patient on infliximab and adalimumab.

Details on the previous biological treatment in the study group (biologic-naïve patients had not been treated with biologic drugs before, biologic-exposed patients received at least one dose of a biologic for UC prior to study enrollment, biofailure patients failed treatment due to lack or loss of response or treatment intolerance).
Clinical characteristics and comorbidities
Most patients (n = 54, 54%) in the total study population were diagnosed with UC 6 or more years prior to study enrollment (Table 3). In the subpopulation of biologic-naïve patients, disease duration was significantly shorter than in either the biologic-exposed or biofailures subgroups. The median (IQR) duration of UC was 6 (3–11) years in the total study population, 5 (2–10) years in biologic-naïve, 8 (5–12) years in biologic-exposed, and 7 (4–11) years in biofailure patients. Extensive colonic involvement (E3) was present in approximately half of the patients, and more than 40% of patients had the disease limited to left-sided colitis in the total study population and in each subgroup. Approximately one-third of the patients in the total study population and in each subgroup was diagnosed with extraintestinal manifestations of UC during the course of the disease, with arthralgia and aphthous stomatitis being the most common. At study enrollment, any extraintestinal manifestation of UC (mainly arthralgia) was reported in approximately 13% of patients in the overall study population. Details on patient clinical characteristics are shown in Table 3.
Clinical characteristics of the study population.

E1, ulcerative proctitis; E2, left-sided ulcerative colitis; E3, extensive ulcerative colitis; EIM, extraintestinal manifestation; IQR, interquartile range.
biologic-naïve vs biologic-exposed.
biologic-naïve vs biofailures.
U Mann–Whitney test.
Fisher test.
available for 98 patients (54 biologic-naive and 44 biologic-exposed).
Hospitalization due to exacerbation of UC can be an indicator of disease activity and severity and can have an impact on prognosis. Approximately two in three patients (n = 68, 68%) were hospitalized due to worsening UC in the 12 months prior to enrollment: 47 (47%) patients were hospitalized once, 16 (16%) patients twice, and 2 (2%) and 3 (3%) patients reported 3 and 4 hospitalizations, respectively. The mean (standard deviation) total duration of hospitalization was 14.2 (17) days.
Disease activity at enrollment, assessed with the Mayo scale, was generally similar among analyzed subgroups (Table 4). In the overall population, the median (IQR) Total Mayo Score was 10 (9–11), and the median Partial Mayo Score was 7 (6–8). No significant differences in the median Mayo scores and in the Mayo subscales were observed between the biologic-naïve and biologic-exposed or biofailure subgroups. Approximately three in four patients in the overall study population as well as in each subgroup reported more than 4 stools a day more than normal. Most patients in each subgroup reported visible blood in stool at least half of the time, and pure blood was reported in 14% of individuals from the total study population and in 20% of biologic-naïve patients. Upon endoscopy, mucosal appearance indicated severe disease activity in approximately two-thirds of patients and moderate activity in approximately a quarter of patients from the overall population. All UC patients were classified as moderately or severely active upon the global assessment by a physician.
Baseline disease activity according to the Mayo Score.
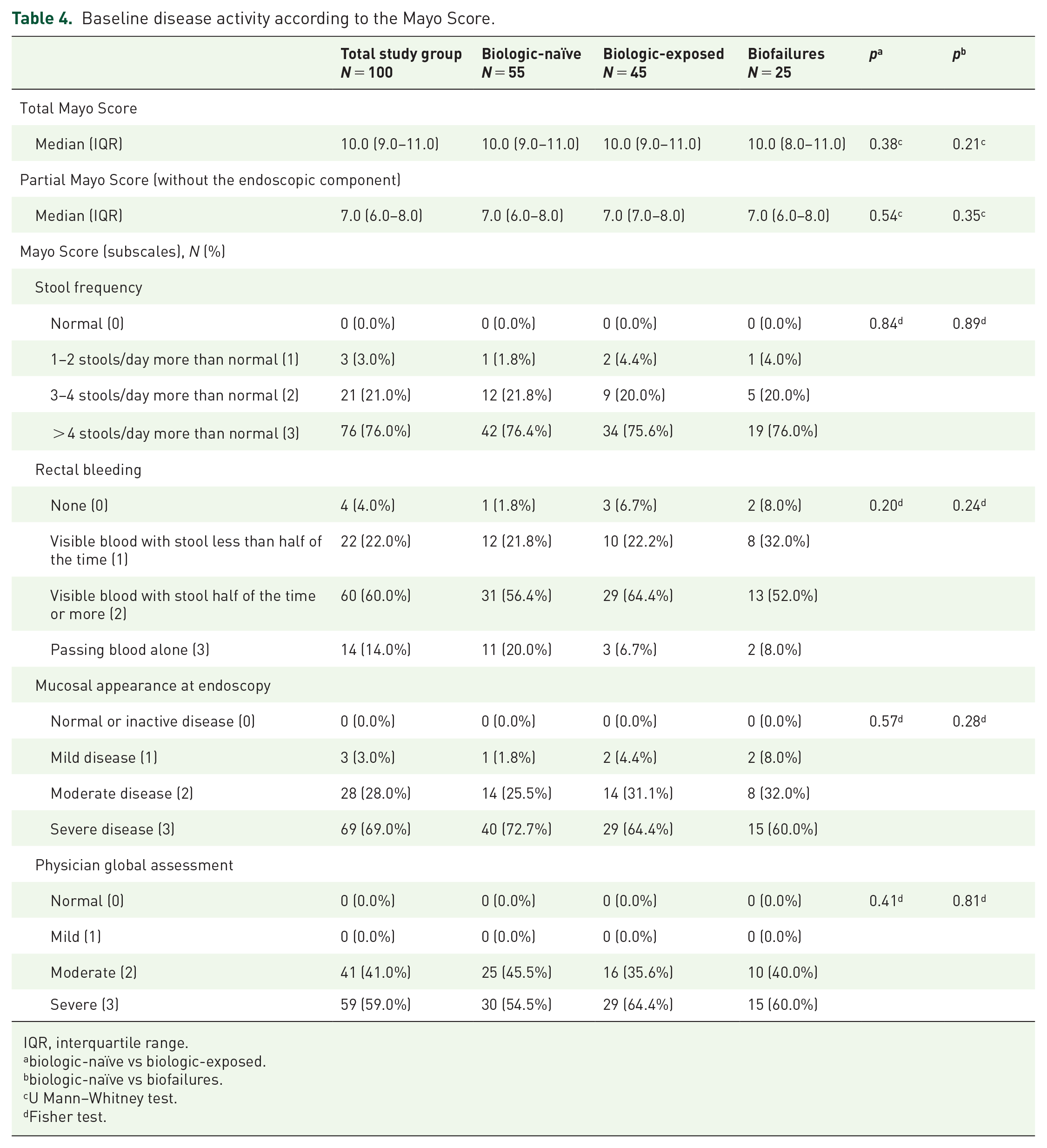
IQR, interquartile range.
biologic-naïve vs biologic-exposed.
biologic-naïve vs biofailures.
U Mann–Whitney test.
Fisher test.
Comorbidities affected 27 (27%) patients and included autoimmune disorders such as autoimmune hepatitis, Graves’ disease, coeliac disease, rheumatoid arthritis, and psoriasis (Table 5). Ten (10%) individuals reported 2 or more comorbidities. Diabetes mellitus, obesity, and hypertension were the most commonly reported comorbidities.
Comorbidities.

including cysts and polyps.
Concomitant treatment
In the total study population, 68 (68%) patients received concomitant systemic corticosteroids, mostly methylprednisolone and prednisone (Table 6). The median dose equivalent of prednisolone was 20 mg daily. No significant differences were observed in the median daily dose of prednisolone between the subgroups. Overall, more than half of the patients were corticosteroid-dependent, and approximately one in five patients were corticosteroid-refractory. A significantly lower percentage of biologic-naïve than biologic-exposed or biofailure patients was corticosteroid-dependent. By contrast, corticosteroid refractoriness was significantly more prevalent in biologic-naïve than in biologic-exposed and biofailure individuals. Approximately 5% of patients had corticosteroid intolerance in the total study group and in all subgroups. Immunomodulator treatment was used in nearly half of the patients. The most common immunomodulator taken was azathioprine, and the median daily dose was 100 mg. Among 5-ASA derivatives, most patients were treated with mesalazine. The profile of concomitant treatment with immunomodulators was generally similar among different biologic treatment subgroups.
Baseline non-biologic treatment.

biologic-naïve vs biologic-exposed.
biologic-naïve vs biofailures.
U Mann–Whitney test.
chi-square test.
Discussion
Here, we report the detailed demographic and clinical characteristics of patients enrolled in the POLONEZ study, who started treatment with the selective integrin antagonist vedolizumab within the scope of the NDP. In Poland, the access to treatment with vedolizumab is limited to the NDP, which not only restricts treatment availability to approved, highly specialized centers, but also implies strict inclusion criteria. 18 This influences the characteristics of the patient population, which may differ from other real-word patient populations across Europe, and justifies a need to collect treatment outcomes.
The onset of UC usually occurs between the age of 30 and 40 years. 2 A similar age profile of UC patients in clinical practice was shown in real-world studies.17,24 The population of patients included in our study tended to be younger compared with those included in most RWE studies on vedolizumab. The reported median disease duration of 6 years indicates that most patients in our study population had been diagnosed with UC under the age of 30 years. In most real-world studies conducted across Europe, the duration of UC among patients treated with vedolizumab was longer compared with that reported in our study. The median time from diagnosis was 10 years in a study conducted in a Spanish25 cohort and 7 years in a German study. 26 In a French vedolizumab cohort, the mean duration of disease was 8.8 years. 27 Like in our study, a shorter disease duration was reported in studies with vedolizumab-treated patients in Scotland (6 years)28 and in Sweden (4 years). 29
In our study population, the baseline median Total and Partial Mayo scores of 10 and 7, respectively, indicate that patients had severely active UC upon enrollment. In the GEMINI 1 trial, the mean values of Total and Partial Mayo scores were 8.6 and 6.0, respectively. 16 In line with the GEMINI 1 trial, a median Partial Mayo score of 6 was reported in real-world studies conducted in Spain,25 Scotland,28 and Germany. 26 This is consistent with the European Medicines Agency (EMA) label stating that vedolizumab is indicated for the treatment of moderately to severely active UC. 19 Furthermore, approximately two-thirds of patients in our study population were hospitalized due to UC worsening within 12 months prior to study enrollment, while less than one third of patients required hospitalization for UC worsening in studies conducted in Germany (27%)26 and Scotland (34%). 28 Taken together, these data indicate a more severe disease activity in the Polish study population compared with corresponding European cohorts treated with vedolizumab.
Disease extent in patients receiving vedolizumab was similar across European real-world studies, with approximately 54–69% of patients affected by extensive UC.25–30 In our study, extensive colitis was observed in approximately half of the patients. Extraintestinal manifestations were reported in one-third of our study population, and one in eight patients experienced extraintestinal symptoms at study enrollment. Similar percentages of patients reporting such symptoms were observed in other real-world populations receiving vedolizumab treatment.25–28
The effectiveness of vedolizumab for UC was generally higher among patients who had not been previously treated with anti-TNF drugs than in those who had been.17,28,31,32 In most real-world studies addressing outcomes of vedolizumab therapy, most patients had been previously treated with anti-TNF drugs. Two large meta-analyses assessing the effectiveness of vedolizumab for inflammatory bowel disease in a clinical practice setting showed that anti-TNF-naïve patients constituted on average 14.5% (for UC only)24 and 19.6% (UC and Crohn’s disease (CD) patients combined)17 of the total population studied in real-world studies. In studies conducted across Europe, only 2.4% of patients were anti-TNF-naïve in a French population,27 whereas 24.3% of such patients were reported in Germany. 26 In a Scottish study, Plevris and colleagues28 highlighted a large number of anti-TNF-exposed patients as one of the main limitations of available real-world studies and reported a total of 38.3% of anti-TNF-naïve UC patients in their study population. By contrast, in our study, approximately half of the patients were biologic-naïve. Many of these differences in study populations across Europe might result from different local or national reimbursement policies across the countries; the NDP in Poland offers both infliximab and vedolizumab as first-line biologic treatment options. Hence, the ongoing assessment of the treatment outcomes in our study population appears to be relevant for an optimal positioning of vedolizumab in the treatment of UC in Poland.
In our study, approximately half of the patients had previously failed to respond to at least one biologic treatment of UC. Most studies investigating vedolizumab for the treatment of UC report a much higher percentage of patients who failed previous biologic treatment. In the GEMINI 1 trial, 85% of enrolled patients previously treated with anti-TNFs were reported as failures,16 and in the VARSITY trial, 94% of patients had a documented failure to previous anti-TNF treatment. 31 In a real-world population receiving vedolizumab for UC in Germany, a failure to respond to previous biologic treatment was reported in 98% of biologic-exposed patients. 26 Such a discrepancy in the percentage of baseline biofailure individuals between our study and other studies most likely results from the Polish reimbursement system for UC biologic treatment that mandates patients on a biologic be taken off the respective biologic treatment following a maximum of 52 weeks for infliximab and 54 weeks for vedolizumab, irrespective of their disease being in clinical remission or not. 18 However, the Polish reimbursement criteria allow for a restart of the biologic treatment provided a patient experiences a disease flare classified as moderately to severely active. Furthermore, up to mid-2018, infliximab was the only biologic reimbursed, and if patients failed (non-response, loss of response, or intolerance) on infliximab, no other option was available to them.
The frequency and type of concomitant medication use in patients treated with vedolizumab can differ across Europe. Usage of both immunomodulators and corticosteroids in the Polish population described in this study corresponds to the average usage of these medications shown in clinical trials and real-world studies on vedolizumab. In the GEMINI 1 trial, 34% of the patients were treated with immunomodulatory drugs and 54% with corticosteroids. 16 According to a meta-analysis by Engel and colleagues,24 including 9 real-world studies on vedolizumab-treated patients in the United States, Europe, and Asia, overall 56% of UC patients were co-treated with immunomodulators and 59% with corticosteroids. Immunosuppressants were used in as much as 76% of the patients from a German26 cohort, and in 65% of a Spanish real-world cohort. 25 In studies from Scotland and France, the concomitant use of immunosuppressants was less frequent and involved only 32% and 22% of patients, respectively.27,28 Real-world studies from Europe show that most patients receive concomitant treatment with corticosteroids (from 44% in France to 83% in Germany).25–28
The percentage of UC patients developing corticosteroid dependence or refractoriness reported in real-world studies varies. According to Faubion and colleagues,33 22% of UC patients treated with corticosteroids were corticosteroid-dependent and 16% were corticosteroid-refractory (defined as no response 30 days after the introduction of corticosteroid therapy). In a study by Ho and colleagues, 34 corticosteroid dependence and refractoriness was reported in 17% and 18% of patients, respectively. A more recent study, including a group of 464 patients treated with corticosteroids, showed that 38% of patients were corticosteroid-dependent and 11% were corticosteroid-refractory. 35 Corticosteroid dependency seems to occur more frequently in patients who receive corticosteroid treatment early on in the course of the disease (i.e. within 30 days after UC diagnosis) than in those who required no such intervention,36 which likely indicates severe disease activity. In corticosteroid- dependent or refractory UC patients, it is considered appropriate to switch to an immunosuppressive or biologic therapy in order to control the disease and avoid the well-known side effects of corticosteroids. 37 The population included in our study followed the inclusion criteria of the NDP and, therefore, mainly consisted of patients in whom immunosuppressive treatment with azathioprine or mercaptopurine had failed, and corticosteroid dependency was the main indication to initiate a biological therapy. Therefore, the inclusion criteria of the NDP can explain the high percentage of patients with corticosteroid dependence in our study.
The differences in baseline characteristics between populations treated with vedolizumab in real-world settings may be attributed to an unequal access to biologic treatment among eligible patients. Physicians’ and patients’ preferences, limited access to healthcare and specialists, and delayed diagnosis are mentioned as barriers that restrict access to biological treatment for UC. 38 Differences in the availability of such treatment in particular countries and regions are most likely driven by financial reasons.38,39 Indeed, a large discrepancy exists in the access to biologic treatments for inflammatory bowel disease (authorized by the EMA) across Europe.5,38,39 According to Péntek and colleagues,38 out of the 10 European countries included in their analysis, the estimated percentage of patients with CD treated with biologics was the highest in France (31.3% of patients receiving biologic treatment), followed by Spain (25%), Hungary (19.1%), Slovakia (18.7%), Sweden (15.4%), Germany (15%), and Czech Republic (11.3%). By contrast, in Poland, Romania, and Latvia, access to such treatment was severalfold lower (2.8%, 2.3%, and 0.2% for Poland, Romania, and Latvia, respectively). 38 In Poland, the main barriers to biologic treatment in CD include limited drug availability due to financial reasons, physicians’ preferences, strict reimbursement criteria, and limited access to specialized centers and healthcare in general. 38 Access to biologic treatment for UC and other immune-mediated inflammatory diseases in Central and Eastern Europe (CEE) is generally lower than in Western European countries. 39 Examples of different eligibility criteria for treatment with biologic drugs are presented in Table 7. Among the seven analyzed countries, requirements for access to biologic treatment were the strictest in Poland and Bulgaria. In the five included countries, biologics were indicated for patients with a Mayo Score > 6. Failure (or intolerance) to both corticosteroids and immunomodulators is a requirement to initiate biologic treatment in Poland, Latvia, the United Kingdom, and France (Table 7). Within CEE, the percentage of patients with UC on biologic therapy varies. According to a 2015 study by Rencz and colleagues,5 the estimated percentage of UC patients treated with biologics in the CEE region was highest in Slovakia (6.4%), followed by Hungary (3.5%), Romania (2.1%), Estonia (1.3%), and Lithuania (1%), and the treatment was unavailable in Latvia and Bulgaria.
Eligibility criteria for the treatment of ulcerative colitis with biologic therapeutics in selected European countries based on literature data.

GP, general practitioner.
Data were taken from Polish Ministry of Health18 for Poland, Bortlík and Collection of Laws of the Czech Republic40–42 for Czech Republic, The National Health Service of the Republic of Latvia43 for Latvia, National Health Insurance Fund44 for Bulgaria, Directorate-General for Health,45 Diário da República Eletrónico, 46 and Government Directive47 for Portugal, National Institute for Health and Care Excellence48 for UK, and Légifrance and Agence Nationale de Sécurité du Médicament et des Produits de Santé49–51 for France. Modified and updated from Péntek and colleagues.38
At the discretion of a particular center.
For example, approval or authorization by the health insurance fund, approval of specialists’ Concilium.
The higher the score, the stricter the eligibility criteria in the country.
This study has certain limitations. Due to the diverse inclusion criteria for treatment of UC with biologics globally and across Europe, data gathered for the population within the POLONEZ study can refer only to the Polish population. Nevertheless, such an approach allows us to assess the effectiveness and safety of vedolizumab in a specific group of patients, defined by the NDP prerequisites. We put our results into the context of data obtained in other countries based on the available literature data only. Different eligibility criteria for treatment with vedolizumab as well as the lack of direct access to raw data on the characteristics of the other European populations make the comparative statistical analysis impossible to conduct because of the potential numerous confounding factors. Moreover, as a real-world study, our analysis typically could be affected by less rigorous data collection than those obtained in randomized controlled trials. However, in our study, the strict reimbursement regulations required the apatient characteristics to be thoroughly examined and documented in a uniform manner, allowing for a complete clinical profile of all enrolled patients. Importantly, despite the limitations, real-world studies can provide information on a specific population relevant to clinical practice as compared to randomized controlled trials, which tend to exclude certain subgroups of patients and therefore, often address only a selected group of the total patient population. Nationwide registries that collect data from large cohorts of UC patients contribute considerably to better management of the disease. In Poland, however, such a detailed registry is not currently available. Therefore, the POLONEZ study, which includes a relatively small population of 100 patients, provides meaningful data on patient characteristics and the effectiveness and safety of patients treated with vedolizumab in the context of the NDP, considering local clinical practice that may allow an extrapolation to the overall Polish UC population treated with vedolizumab.
To conclude, in this study we provide detailed baseline characteristics of patients who started treatment with vedolizumab in the context of the NDP in Poland. The Polish population seems to be distinct from those described in other real-world cohorts of vedolizumab-treated patients across Europe, especially in terms of a higher percentage of patients with a more severe disease activity and a higher proportion of biologic-naïve patients. In addition, it can be hypothesized on the basis of literature data that patients in Poland tend to receive vedolizumab treatment earlier after UC diagnosis than those from other European cohorts. Further analysis of treatment effectiveness and safety in this population might contribute to the ongoing discussion on the appropriate positioning of vedolizumab in the management of UC and, in particular, to a better understanding of its potential benefits as a first-line biologic treatment after conventional therapy.
Footnotes
Acknowledgements
The authors thank the following persons for their contributions to data collection, analysis, assistance with statistical analysis, critical review of the article, and medical writing support: (1) National Cancer Institute: Monika Cichaczewska; (2) Święcicki University Hospital: Agnieszka Dobrowolska, MD, PhD, Anna Królewska; (3) Takeda Pharma Sp.z.o.o.: Magda Zbrzeźniak, Paulina Batóg, Monika Smałz (4) Biostat: Edyta Klemba, Marian Płaszczyca, PhD, Barbara Gorzawska; (5) Proper Medical Writing sp. z o.o.: Maria Kołtowska-Häggström, MD, PhD, Anna Woziwodzka, PhD, Ewa Marczuk, funded by Takeda Pharma Sp.z.o.o.
Author contributions
E.Z. and K.W. conceived the concept of the study. E.Z. and K.W. contributed to the design of the study. All authors were involved in data collection. S.D. and K.W. analyzed the data. S.D. coordinated the project and funding for the project. All authors contributed to the manuscript and approved its final version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by Takeda Pharma Sp.z.o.o.
Conflict of interest statement
Piotr Eder received lecture fees and/or travel grants from Takeda, Ferring, Astellas, Pfizer, and Janssen. Kamila Stawczyk-Eder received travel grants and lecture fees from Janssen, Pfizer, and Takeda. Renata Talar-Wojnarowska received lecture fees and/or travel grants from Abbvie, Astellas, Ferring, Janssen, and Takeda. Hubert Zatorski gave scientific advice to Takeda. Anna Solarska-Półchłopek received lecture fees and travel grants from Janssen. Rafał Filip served as a speaker for Gramineer International AB, Egis, Ferring, Janssen, and Takeda; received investigational grants from Gramineer International AB, and Egis; and received support for traveling and congress assistance from MSD, Abbvie, Egis, Takeda, and Ferring. Maria Kłopocka has received payment for lectures from Janssen, Takeda, Ferring, Alfa-Sigma, and Pharmabest and travel/accommodation/meeting expenses from Ferring, Janssen, Takeda, Alfa-Sigma, and Pharmabest. Ariel Liebert received payment for lectures from Janssen, Takeda, Egis, Abbvie, and Pharmabest and travel/accommodation/meeting expenses from Janssen, Takeda, Egis, and Abbvie. Aleksandra Kaczka received lecture fee(s)/travel/accommodation/meeting expenses from Takeda, Janssen‑Cilag, Biogen, Astellas, and Alfa-Sigma. Krzysztof Wojciechowski and Szymon Drygała are permanent employees of Takeda Pharma Sp.z.o.o. Edyta Zagórowicz received lecture fees from Janssen, Sandoz, Ferring, and Pfizer; consultancy fees from Pfizer, Janssen, and Takeda; and other compensations from Takeda and Janssen. The remaining authors disclose no conflicts of interest.