
Abstract
Background:
Active disease during conception and pregnancy in women with inflammatory bowel disease (IBD) increases the risk of pregnancy complications and adverse neonatal outcomes. The use of IBD treatments during pregnancy should be weighed against their adverse effects on the neonate, but longer-term safety data and data on serious infection rates and malignancies postnatally are lacking, particularly for newer drugs, such as tofacitinib, vedolizumab and ustekinumab.
Methods:
This ongoing, prospective registry study being conducted at 70 centres in Spain is enrolling pregnant women who are ⩾18 years, are at any point in pregnancy up to the end of the second trimester and have a diagnosis of Crohn’s disease, ulcerative colitis or unclassified IBD. Patients will receive treatment decided independently by their IBD specialist. Each incident gestation will be followed up through pregnancy and the first 4 years postnatally. Three cohorts will be compared: biologicals exposed, immunomodulatory exposed and non-exposed. The primary endpoint is the risk of severe infection in newborns postnatally up to 4 years of age; other endpoints include serious adverse events (SAEs) such as pregnancy and delivery complications, neonatal SAEs, development [Ages and Stages Questionnaire-3 (ASQ3)], and malignancy incidence, up to 4 years of age. IBD specialists will collect maternal data (baseline/end of each trimester/1 month post-delivery), neonatal birth data, and the SAE and ASQ3 data in children exposed during pregnancy, reported every 3 months by the mother. Statistical analysis will include summary statistics for quantitative variables, comparisons of qualitative variables with significance set at p < 0.025 and a binary logistic regression model to determine the risk factors for severe infections.
Results:
Enrolment began in September 2019 and study completion is expected in September 2028.
Conclusions:
This prospective, controlled study will provide evidence on the long-term safety profile in children after intrauterine and lactation exposure to biological and immunomodulatory IBD treatments, including data on postnatal severe infections, development and malignancies.
ClinicalTrials.gov identifier:
NCT03894228
Keywords
Introduction
Inflammatory bowel disease (IBD) affects many female patients of childbearing age. 1 Women with IBD who would like to become pregnant are faced with several concerns, such as the influence of their disease on the foetus and whether their IBD treatment(s) will affect their chances of conception, the foetus during pregnancy, and their baby when breastfeeding.1,2
IBD does not increase the risk of pregnancy complications per se; disease activity is the main factor associated with increased risk of pregnancy complications. 3 Numerous studies have shown that active disease is associated with poor obstetric or neonatal outcomes, including spontaneous abortion,4,5 low birth weight,6,7 small for gestational age 6 and preterm birth.4,6
Indeed, if a patient with IBD conceives when her disease is in remission, the risk of flare-ups is similar to that of non-pregnant women, 8 but if the woman conceives during a period of active disease, there is an eight-fold greater risk of disease flare-up during pregnancy, 9 and a two-fold greater risk of the disease remaining active throughout the pregnancy compared with a patient in remission at the time of conception. 10 Effective disease control both prior to and during pregnancy is therefore critically important. 11 Treatment should be optimised to reduce the risk of IBD flare-up, but any risks associated with continuing IBD treatment before and during pregnancy should be taken into account.12–14
There is evidence that most drugs are safe for use during pregnancy and lactation, although methotrexate and thalidomide are contraindicated.12–14 However, few studies to date have provided long-term follow up of children exposed to IBD drugs. 1
Biological drugs have increasingly been used in the treatment of IBD. The majority of evidence on their use during pregnancy is available for the anti-tumour necrosis factor (anti-TNF) agents infliximab and adalimumab, which appear to be associated with a low risk of adverse outcomes.13,14 There is a lack of evidence for the use of newer biological agents (such as vedolizumab and ustekinumab) and JAK inhibitors (such as tofacitinib) for IBD during pregnancy and lactation, and their long-term effect on children exposed to them in utero. 1 Most clinical trials of these newer agents excluded pregnant women, and those that did include pregnant women had only a small number of patients and a short follow-up period (usually only up to the time of childbirth). 1
Therefore, there is a need for a prospective register that will provide information on the safety of IBD drugs, both well-established and newer agents, in the mother and her exposed child, up to and beyond the first year of age. The aim of the Safety of IB
Methods
Study design and population
DUMBO is a prospective, observational registry that will enrol pregnant women with IBD [either Crohn’s disease (CD), ulcerative colitis (UC) or unclassified IBD] over a 5-year period at 70 centres across Spain [ClinicalTrials.gov identifier: NCT03894228].
Recruitment began in September 2019 and will end in September 2024, with an expected study completion date of September 2028. Registration of the patient and obtaining patient permission is performed by the IBD specialists at each participating centre. Study investigators are responsible for data collection. Each incident gestation will be followed up through pregnancy and during the first 4 years of the child’s life. Treatment and any other interventions are decided independently by the treating physician and according to local clinical practice.
Study population
Female patients aged ⩾18 years with an IBD diagnosis and confirmed pregnancy, with the pregnancy known to the study researcher prior to gestation week 28, are being included in the study. Patients are excluded if they do not consent to participate in the study.
These female patients, as well as the children born to them will comprise the study population, which will be classified into three cohorts as follows: (a) biological-exposed cohort: children born to mothers treated with biological agents (with or without immunomodulatory agents) at any time during pregnancy or the 3 months prior to conception; (b) immunomodulator-exposed cohort: children born to mothers treated with immunomodulatory agents only, at any time during pregnancy or 3 months prior to conception; or (c) non-exposed cohort: children born to mothers with IBD treated with neither biological nor immunomodulatory agents at any time during pregnancy and in the 3 months prior to conception. Children will be assigned to these cohorts based on the treatment (or lack thereof) their mother was receiving at the time of conception (i.e. before study entry). Pregnancy outcome data will also be collected.
Study objectives
The primary study objective is to determine the risk of severe infections in the child from birth to 4 years of age. Other study objectives include establishing the risk of serious adverse events (SAEs) during pregnancy and delivery associated with drugs used to treat IBD, assessing developmental status of the child during the first 4 years of life, comparing the risk of SAEs in children exposed in utero to drugs used to treat IBD with the risk in children who were not exposed, comparing the prevalence of congenital abnormalities in children exposed in utero to drugs used to treat IBD with the prevalence in children who were not exposed, and evaluating the risk of developing neoplasm(s) in children exposed in utero to drugs used to treat IBD compared with children who were not exposed.
Study definitions
IBD location and phenotype will be classified as per the Montreal classification 15 and IBD activity measured using the Harvey–Bradshaw Index for CD 16 and the partial Mayo Score for UC. 17 IBD treatment is considered as treatment received by the mother in the 3 months prior to conception, during pregnancy and during lactation. SAEs are defined as AEs leading to death, a life-threatening event, hospitalisation, disability or permanent damage, congenital anomaly or birth defect, and other AEs that the investigator felt may require intervention to prevent one of the other outcomes. Specific maternal SAEs of interest include abortion, stillbirth, growth retardation, severe infection, eclampsia, placenta praevia, chorioamnionitis or abruptio placentae, foetal malformations and any SAEs during delivery (e.g. instrumental delivery). Neonatal and childhood SAEs of interest include severe infections, congenital malformations, admission to the intensive care unit, low birth weight, hypoxic–ischaemic encephalopathy, neonatal stroke or low Apgar score and tumours (among others). A complete list of study definitions is provided in Table 1.
Study definitions.
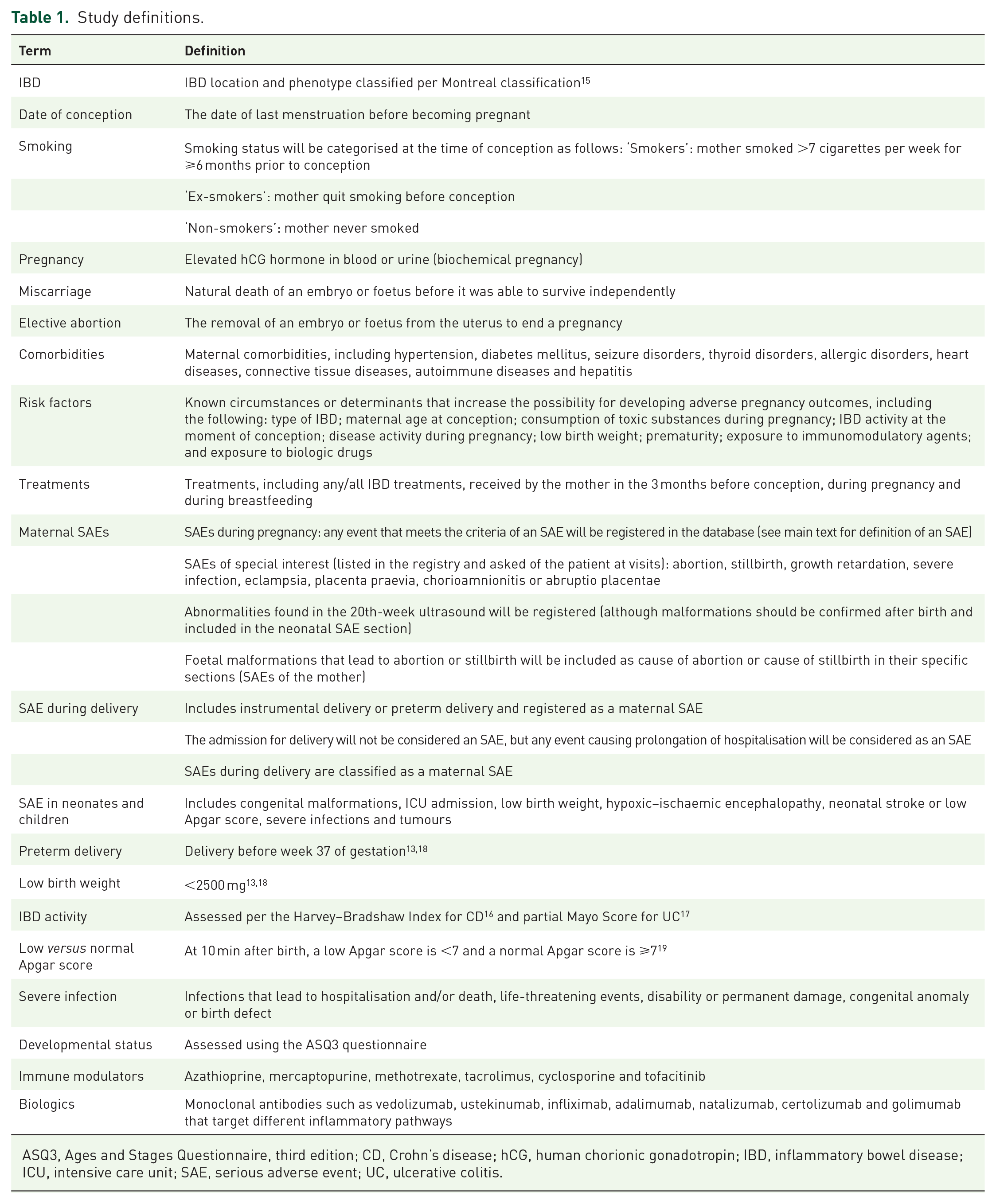
ASQ3, Ages and Stages Questionnaire, third edition; CD, Crohn’s disease; hCG, human chorionic gonadotropin; IBD, inflammatory bowel disease; ICU, intensive care unit; SAE, serious adverse event; UC, ulcerative colitis.
Data collection and follow up
Maternal demographic and clinical data (disease characteristics, activity and treatment) will be collected at baseline (visit 0), defined as the time of study enrolment (after confirmation of pregnancy but before the end of the second trimester). At the end of each trimester and 1 month after delivery (visits 1–4) the following data will be collected: disease activity; treatment(s); and SAEs during pregnancy and delivery. In addition, mothers will be asked to provide information about whether or not they are breastfeeding, and any concomitant medications they took while breastfeeding at each 3-month visit after birth. All data will be collected prospectively.
Each live birth will be registered in the database as a case at visit 4 (1 month after delivery), and the following clinical information will be recorded: date of birth; sex; birth weight; Apgar score at 5 min and 10 min; vaccinations; and SAEs. SAE and developmental status data pertaining to the neonate/child will be reported by the mothers every 3 months after birth; this information will be confirmed annually by medical record review. A study investigator from the coordinating group will contact the mother for completion of the Ages and Stages Questionnaire (ASQ3) every 2 months (age 2–24 months), at 9 months, then every 3 months (age 27–36 months), at 42 months and 48 months of age, and answers will be registered with their IBD specialist by the coordinating centre on a yearly basis.
All study data will be collected by the IBD specialists using the electronic data capture software tool REDCap (Research Electronic Data Capture; Vanderbilt University, Nashville, TN, USA), 20 which is hosted by the Asociación Española de Gastroenterología (AEG, Madrid, Spain), a not-for-profit medical society.
Data verification
Data entered in the registry will be verified by the investigators (MGD, DA, YB) from the Hospital Universitario de La Princesa (Madrid, Spain), who will conduct a review of all cases.
Statistical analysis
Sample size
In order to ensure sufficient statistical power, the study sample size was calculated based on an estimated severe infection rate of 3.2% per person-year of follow up, which is two-fold higher than the rate observed in our previous study with children not exposed to anti-TNF treatment born to mothers with IBD (1.6% per person-year of follow up). 21 Given an estimated loss to follow up of 5%, for 80% statistical power at a significance level α of 5%, the required sample size was calculated to be 1500 person-years per cohort, which equates to a total of 375 children per cohort (exposed to biologics and non-exposed) and followed for 4 years. Therefore, data will need to be collected on a minimum of 750 children born to IBD mothers.
Descriptive statistics will be used for quantitative variables (mean, standard deviation, median and interquartile range) with comparisons made using the Student’s t test. The chi-squared test and/or Fisher’s exact test will be used to compare qualitative variables to derive statistical significance for the overall three-way cohort comparison. If significant, comparisons will be made between each exposed cohort with the non-exposed cohort with significance set at p < 0.025 after adjustment by Bonferroni correction for multiple comparisons.
A binary logistic regression model will be used to determine risk factors for severe infections. Factors both statistically significant and of clinical relevance in the univariate analysis will be incorporated in the multivariate analysis, where the dependent variable will be the presence of severe infections. Independent variables will include: type of IBD; maternal age at conception; consumption of toxic substances during pregnancy; IBD activity at the moment of conception; activity disease during pregnancy; low birth weight; prematurity; exposure to immunomodulatory agents; and exposure to biological drugs.
Ethics
This study protocol was approved by the Research Ethics Committee of Hospital Universitario de La Princesa (Madrid, Spain) and by the Spanish Medicines Regulatory Authority [Agencia Española de Medicamentos y Productos Sanitarios (AEMPS): EPA-AS code: GET-VED-2018-01].
As per current pharmacovigilance legislation, investigators are asked to report any suspected adverse reactions recorded in the study via the web page of the national reporting system [Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (www.notificaRAM.es)].
The study will be conducted in accordance with the 2013 version of Declaration of Helsinki, and according to current Spanish legislation. Patients or their legal guardians will provide written informed consent to participate in the study in accordance with the International Conference on Harmonisation’s Guidelines for Good Clinical Practice and local legal and administrative regulations. Patient identity will remain unknown to the study sponsor; the principal investigator is responsible for maintaining patient confidentiality.
Discussion
The aim of the DUMBO registry study is to address the significant gaps in knowledge regarding longer-term outcomes of children born to mothers receiving IBD treatment during pregnancy and lactation. The study population will be grouped by type of IBD treatment into two cohorts, the biologicals exposed and immunomodulator exposed, and compared with each other and with a reference cohort of children exposed to neither of these drugs. The novelty of this registry study will be in obtaining long-term safety data on IBD treatment in pregnancy, particularly with regard to newer biological and immunomodulatory agents. We asked the centres included in this study about their ability to recruit eligible pregnant patients, and according to their responses, we estimate that we will be able to include data from more than 1500 pregnancies. So far, we have already collected data from 420 pregnancies in the first 18 months of the study. After completion, our registry with 1500 pregnancies will compare favourably to all other registries we are aware of.
Previous studies have frequently examined the effect of IBD treatment exposure in utero on pregnancy outcomes, including abortion, stillbirth, growth retardation and preterm birth, and on neonatal outcomes, such as Apgar scores, congenital malformation and low birth weight. However, data on severe infection rates, tumours and developmental milestones in infants beyond 1 year of age are very limited, particularly for newer agents like tofacitinib, vedolizumab and ustekinumab.
Immunomodulatory agents, such as thiopurines, carry a low risk of adverse pregnancy, neonatal or infant outcomes,22,23 and in utero thiopurine exposure is not associated with changes in neurocognitive development or physical or social health. 24
Anti-TNF biological agents are generally considered safe for use during pregnancy.1,25 A meta-analysis of pregnant women with UC or CD found no increase in the risk of adverse pregnancy outcomes with anti-TNF therapy versus disease-matched controls. 26 Limited longer-term data suggest in utero exposure to anti-TNF therapy had no adverse effects on postnatal development or malignancy risk. In the Pregnancy in Inflammatory Bowel Disease and Neonatal Outcomes (PIANO) registry study (which collected data in approximately 1000 children born to IBD mothers), developmental outcomes up to 1 year of age were unaffected by in utero anti-TNF exposure. 27 Bortlik et al. 28 conducted a study with longer follow up of 25 children exposed to infliximab or adalimumab in utero; these children were followed for up to 70 months (5.8 years), and their growth and psychomotor development was normal in most cases. None of these studies reported data on malignancies; in the long-term, retrospective TEDDY study, there were no malignancies reported in 388 children who were exposed to anti-TNF therapy in utero. 21
The short-term risk of infection does not appear to be increased among children exposed to anti-TNF therapy in utero. In the retrospective EVASION study of pregnant women with IBD, the risk of infection in the first year of life was not increased in children exposed to anti-TNF therapy in utero compared with those not exposed. 29 However, in the PIANO registry study, an increased risk of infection was observed in children aged 9–12 months exposed to both anti-TNF therapy and thiopurines versus those not exposed to either drug class, 27 suggesting an increased risk of neonatal infection with combination therapy. Bortlik and colleagues reported a high incidence of infant infection following in utero infliximab or adalimumab exposure, but a low rate of severe infection; all infections resolved without sequelae. 28 In the TEDDY study, anti-TNF exposure (with or without thiopurines) was associated with a similar incident rate of infection to that observed in non-exposed children over a postnatal follow-up period of 47 months (~4 years; exposed group) or 68 months (5.7 years; non-exposed group). 21
With respect to the current evidence on the safety profile of vedolizumab in pregnant women with IBD and in their infants, the majority of studies were retrospective and lacked a control group, and did not examine outcomes beyond early infancy. 1 The retrospective, pan-European CONCEIVE study was one of the few to collect data on infection rates and malignancies following vedolizumab exposure in infants up to 1 year of age. 30 Among live-born infants, there were no malignancies, and serious infection rates did not significantly differ between the vedolizumab-exposed cohort, the anti-TNF-exposed cohort or the control cohort. Most pregnancy and neonatal outcomes showed no significant differences between groups; the live infant birth rate was significantly lower in the vedolizumab group than the control group (78% versus 89%; p = 0.038), but this difference was no longer significant when patients with active disease during any stage of pregnancy were excluded. 30 To date, one prospective study has been conducted in pregnant women with IBD who were treated with either vedolizumab, anti-TNF agents or conventional therapy, 31 and vedolizumab appeared to be associated with low risk during pregnancy, although the authors concluded that studies with larger populations and longer follow up were needed to confirm this. 31 A clinical programme of six studies in pregnant women provided further evidence on the safety profile of vedolizumab for infants born to women with IBD, 32 but study population was small and follow up of short duration. 32
Published evidence on the use of ustekinumab during pregnancy and its effect on neonates is even more limited than for vedolizumab. Most pregnancy/neonatal safety data are available for patients with psoriasis, where lower ustekinumab doses are used, and therefore, cannot be extrapolated to patients with IBD. 1 To the best of our knowledge, evidence is limited to five case reports of women with CD treated with ustekinumab during pregnancy.33–37 Pregnancy and neonatal outcomes were uneventful in three of them; however, for the remaining women, there was one case of a miscarriage at 8 weeks’ gestation 33 and one of caesarean section due to spontaneous rupture of membranes at 37 weeks’ gestation. 37 Infant follow-up duration was ⩾1 year in three cases, all reporting normal development.34–36
The majority of safety evidence for tofacitinib in pregnant women and their infants is from studies in patients with rheumatological diseases (e.g. psoriatic arthritis and rheumatoid arthritis). 1 One analysis of tofacitinib interventional studies reported positive pregnancy/neonatal outcomes in women with UC receiving tofacitinib at conception or during pregnancy. 38 All women initiated tofacitinib in the first trimester; no foetal or neonatal deaths nor congenital abnormalities were reported, being the most common outcome a healthy newborn. These results were generally consistent with those observed in the entire tofacitinib clinical programme (i.e. UC, psoriasis, psoriatic arthritis and rheumatoid arthritis); however, the authors concluded that there was insufficient evidence to confirm the safety profile of tofacitinib in pregnant women or their infants, particularly given the small patient numbers, lack of controlled studies, and preclinical data indicating tofacitinib’s foeticidal and teratogenic effects. 38
Anti-TNF agents and the newer biological agents vedolizumab and ustekinumab have been detected in very small quantities in human breastmilk; consequently, breastfeeding while taking any of these agents is likely to be of low risk to the neonate.1,11 However, the currently available evidence does not allow for an unequivocal recommendation to continue treatment with these agents during breastfeeding.1,11
Treatment guidelines recommend that administration of live vaccinations to infants with intrauterine exposure to anti-TNF agents12–14,18 and other biologicals12,14 be delayed to after 6 months of age. Non-live vaccinations may be administered according to standard national immunisation schedules. To improve evidence regarding the tolerability of vaccinations, all relevant data on vaccinations will be captured for infants included in the DUMBO registry.
The DUMBO registry has been designed to address the dearth of evidence from prospective controlled studies on the longer-term effects of in utero exposure to IBD treatments, particularly biological agents, on the health and development of the children born to mothers with IBD. The DUMBO study includes a control cohort, and is prospective, which should yield better accuracy of data collection on treatment exposure, endpoints and confounding variables. 39 The primary endpoint of the DUMBO study is the severe infection rate during postnatal development up to 4 years of age, which will provide valuable data regarding the longer-term severe infection risk following in utero exposure to IBD treatment. Similarly, the registry’s secondary endpoints on long-term risk of SAEs, effects on developmental status, and malignancy rate, as well as monitoring the safety of IBD treatment in children who are breastfed (Table 1), will provide further long-term safety data in children up to 4 years of age regarding the use of biological and immunomodulatory agents during pregnancy in women with IBD. In addition, developmental status will be recorded formally using a standardised, well-recognised metric (ASQ3), which will help reduce recall bias regarding the child’s development. Statistical analyses are planned to minimise selection bias and the effect of confounding variables.
In summary, the DUMBO study will provide clinicians with important information on the short- and long-term risks associated with in utero exposure to biological or immunomodulatory agents in women with IBD and their children. It is expected that the results of the registry will address the need for information on the safety of treatments for IBD during pregnancy and lactation and will contribute on establishing evidence-based recommendations for the use of these agents in pregnant women with IBD.
Footnotes
Acknowledgements
We would like to thank Tracy Harrison of Springer Healthcare Communications for her assistance writing the outline and first draft of this manuscript. This editorial assistance was funded by a grant from Instituto de Salud Carlos III (grant number ICI19/00083).
Author contributions
María Chaparro1, Francisco Abad Santos2,3, Francisco Javier Martín de Carpi4, Miguel Ángel Maciá-Martínez5, Dolores Montero5 and Javier P. Gisbert1: study design.
María G Donday, Diana Acosta, Yanire Brenes: study management, case report form design.
Conflict of interest statement
MCh has served as a speaker, or has received research or education funding from MSD, Abbvie, Hospira, Pfizer, Takeda, Janssen, Ferring, Shire Pharmaceuticals, Dr Falk, Pharma, Tillotts Pharma.
FAS has been consultant or investigator in clinical trials sponsored by the following pharmaceutical companies: Abbott, Alter, Chemo, Cinfa, FAES, Farmalíder, Ferrer, GlaxoSmithKline, Galenicum, Gilead, Italfarmaco, Janssen-Cilag, Kern, Normon, Novartis, Servier, Silverpharma, Teva and Zambon.
JPG has served as a speaker, a consultant and advisory member for or has received research funding from MSD, Abbvie, Hospira, Pfizer, Kern Pharma, Biogen, Takeda, Janssen, Roche, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma, Chiesi, Casen Fleet, Gebro Pharma, Otsuka Pharmaceutical, Vifor Pharma.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was funded by a grant from the Instituto de Salud Carlos III [grant number ICI9/00083]. Co-funded by FEDER funds.