
Abstract
Pancreatic ductal adenocarcinoma (PDAC) accounts for about 3% of all cancers in the United States and about 7% of all cancer deaths. Despite the lower prevalence relative to other solid tumors, it is one of the leading causes of cancer-related death in the US. PDAC is highly resistant to chemotherapy as well as radiation therapy. Current standard-of-care chemotherapeutic regimens provide transient disease control but eventually tumors develop chemoresistance. Tumors that are deficient in DNA damage repair mechanisms such as BRCA mutants respond better to platinum-based chemotherapies. However, these tumor cells can utilize the poly adenosine diphosphate (ADP)-ribose polymerase (PARP) as a salvage DNA repair pathway to prolong survival. Hence, in the presence of BRCA mutations, the inhibition of the PARP pathway can lead to tumor cell death. This provides the rationale for using PARP inhibitors in patients with BRCA mutated PDAC. The phase III POLO trial showed a near doubling of progression-free survival (PFS) compared with placebo in advanced PDAC when a PARP inhibitor, olaparib, was used as maintenance therapy. As a result, the US Food and Drug Administration (FDA) approved olaparib as a maintenance treatment for germline BRCA mutated advanced PDAC that has not progressed on platinum-based chemotherapy. The success of olaparib in treating advanced PDAC opened the new field for utilizing PARP inhibitors in patients with DNA damage repair (DDR) gene defects. Currently, many clinical trials with various PARP inhibitors are ongoing either as monotherapy or in combination with other agents. In addition to germline/somatic BRCA mutations, some trials are enrolling patients with defects in other DDR genes such as ATM, PALB2, and CHEK2. With many ongoing PARP inhibitor trials, it is hopeful that the management of PDAC will continuously evolve and eventually lead to improved patient outcomes.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for about 3% of all cancers in the United States and about 7% of all cancer deaths. 1 Despite the lower prevalence relative to other solid tumors, it is the third leading cause of cancer-related death in the US and by 2030, it is projected to become the second leading cause of death. 2 Only about 20% of PDAC patients are diagnosed with early stage or localized disease that is surgically resectable and potentially curable.3,4 Despite early diagnosis, surgical resection and adjuvant chemotherapies provide modest survival benefit. Most of these patients will have local or distant recurrence and eventually die from PDAC. The current 5-year relative survival rates for localized, regional, and advanced stages are 37%, 12%, and 3%, respectively. 1 The median overall survival (OS) is 6.7–11.1 months for advanced disease and 25–28 months for early-stage disease.5–7 Currently, treatment options for advanced PDAC are limited. First line chemotherapies include 5-fluorouracil (5-FU), leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) or gemcitabine plus nab-paclitaxel. However, despite moderate clinical benefit, PDAC eventually develops resistance to conventional chemotherapies.
PDAC is highly resistant to chemotherapy as well as radiation therapy relative to other solid tumors due to the dense desmoplasia surrounding the primary tumor and complex tumor microenvironment. The pancreatic tumor microenvironment consists of various types of cells such as cancer associated fibroblasts, myeloid derived suppressor cells, and regulatory T cells; the interactions among these cells lead to evasion of immune surveillance, creation of a physical barrier to chemotherapeutic agents, promotion of angiogenesis, invasiveness, and chemoresistance.8–12 The myriad of mechanisms underlying chemoresistance in pancreatic cancer also provide opportunities to explore an array of treatment strategies. Various targeted therapies are being studied in clinical trials to target tumor DNA repair mechanism (PARP), tumor metabolic pathways (mitochondrial inhibitor), focal adhesion kinase, and connective tissue growth factors. The recent success of PARP inhibitors in improving clinical outcomes in advanced PDAC has generated a lot of excitement in the field of pancreatic cancer research. Currently, many clinical trials with various PARP inhibitors are ongoing either as monotherapy or in combination with other agents in patients with PDAC.
Rationale of PARP inhibition
PARP enzymes primarily repair single-strand DNA breaks and play crucial roles in DNA damage repair (DDR).13,14 PARP inhibitors are small molecules that trap PARP enzymes on DNA and prevent the process of DDR. Accumulation of single-strand DNA breaks in the presence of PARP inhibitors results in the formation of double-strand breaks, which require homologous recombination to repair. 15 Cancer cells that harbor mutations in DNA repair genes such as BRCA1, BRCA2, PALB2 or ATM are unable to utilize DNA repair via homologous recombination and accumulate double-strand DNA breaks over time, resulting in cell death 15 (Figure 1). In PDAC, it has been shown that patients with mutations in BRCA1 or BRCA2 have better overall survival (OS) when treated with platinum-based chemotherapy compared with those treated with non-platinum-based therapies. 16 This is due to the inability of tumor cells harboring the BRCA mutation to repair the double-strand DNA breaks generated by platinum-based compounds. However, tumor cells can still leverage the PARP pathway to prolong survival. Therefore, inhibiting PARP can lead to tumor cell death in the presence of mutations in genes related to double-stranded DNA repair, such as BRCA1, BRCA2, PALB2, or ATM.17,18 Several PARP inhibitors have already been approved for the treatment of patients with germline or somatic BRCA mutant breast, ovarian, prostate, and most recently, advanced pancreatic cancer 19 (Table 1).

DNA repair of single-strand break in the presence of PARP inhibitor resulting in double-strand break formation. Cells with intact BRCA have the ability to repair the double-strand break, maintaining cell survival. BRCA mutant cells are unable to repair the accumulating double-strand breaks resulting in cell death.
Food and Drug Administration (FDA) approved PAPR inhibitors and their indications.

FDA, US Food and Drug Administration.
BRCA mutations in pancreatic cancer
A large case–control study of germline investigation of PDAC in 2018 identified mutations in six genes associated with PDAC (CDKN2A, TP53, BRCA1/2, ATM and MLH1) with a prevalence of 5.5%. 20 BRCA1 and BRCA2 are genes that code for tumor suppressor proteins involved in the repair of double-strand DNA breaks via homologous recombination. 21 BRCA1 and BRCA2 were found in 0.6% and 1.9% of PDAC patients, respectively. Patients with PDAC are 2.56-fold more likely to have germline BRCA1 mutations, and 6.2-fold more likely for the BRCA2 mutation relative to general population. Non-Hispanic white individuals made up the majority (95%) of the patient population in this study. 20 In high-risk families with multiple incidences of PDAC, the frequency of BRCA2 mutations is as high as 15%. 22 Of note, in the Ashkenazi Jewish population, up to 21% of patients with PDAC were found to have a BRCA2 mutation. 22
Olaparib in pancreatic cancer
The data for the Pancreas Cancer Olaparib Ongoing (POLO) trial were presented at the plenary session of the American Society of Clinical Oncology (ASCO) 2019 meeting. The phase III randomized, double-blind controlled trial screened a total of 3315 patients with metastatic PDAC in 10 countries. Eligible patients had mutations in germline BRCA1 or BRCA2 and received at least 16 weeks of first line platinum-based chemotherapy, mainly FOLFIRINOX. One hundred and fifty-four patients who met the inclusion criteria were randomly allocated either to olaparib 300 mg twice daily or matching placebo as maintenance therapy, within 4–8 weeks of completion of chemotherapy. Of the 154 randomized, 92 patients were assigned to receive olaparib and 62 patients were assigned to a placebo. The primary endpoint of the study was PFS, which was significantly longer in the olaparib arm than the placebo arm [median PFS 7.4 months versus 3.8 months; hazard ratio (HR) 0.53; 95% confidence interval (CI) 0.35–0.82; p = 0.004]. 23 At the interim analysis, the secondary endpoint, median OS, was 18.9 months in the olaparib arm and 18.1 months in the placebo arm (HR 0.91; 95% CI 0.56–1.46, p = 0.68) but it was not statistically significant 23 (Table 2). Olaparib was well tolerated, with a 5% rate of discontinuation versus 1.7% in the placebo group due to toxicity. Anemia and fatigue were the most common grade 3 or more adverse events and was seen in 39.6% in the olaparib group and 23.3% in the placebo group. 23 There was no change in quality of life compared with the placebo arm as measured by the European Organization for the Research and Treatment of Cancer (EORTC) QLQ-C30 score, which is an important goal for any maintenance therapy.
POLO Trial, Progression Free Survival (PFS) and Overall Survival (OS).
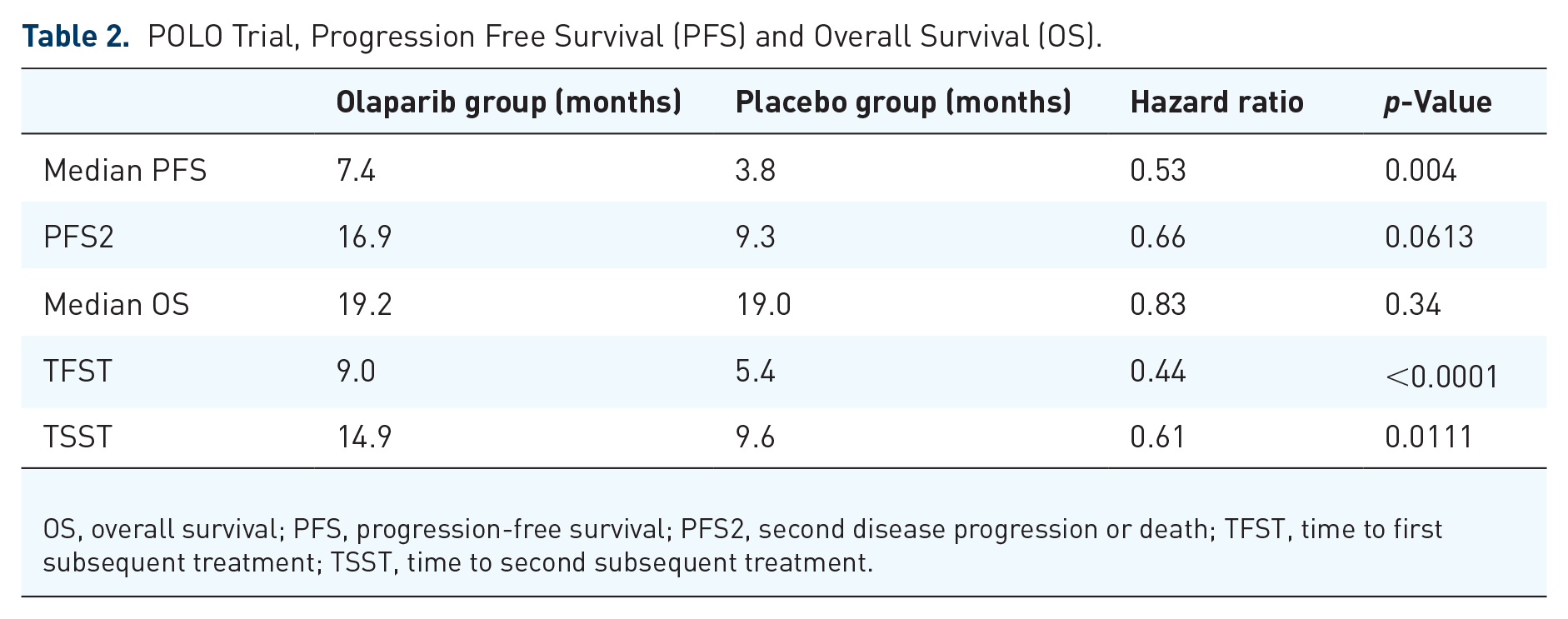
OS, overall survival; PFS, progression-free survival; PFS2, second disease progression or death; TFST, time to first subsequent treatment; TSST, time to second subsequent treatment.
At the annual Gastrointestinal Cancers Symposium in January 2021, the investigators reported an update on the study outcomes. The median OS was 19.2 months for the olaparib group and 19.0 months for the placebo group (HR 0.83; 95% CI 0.56–1.22; p = 0.3487), but it was not statistically significant. 24 It should be noted that 26% of patients in the placebo group received olaparib, and received multiple subsequent lines of therapies on progression of disease and after stopping olaparib, which could bias the OS. Another point to consider is that the POLO study was inadequately powered to detect difference in OS between the two groups. However, there is a larger proportion of patients on olaparib who survived longer than the placebo group after 2 years. Three-year OS was 33.9% for the olaparib group and 17.8% for the placebo group. Another key secondary endpoint, time to first subsequent treatment (TFST), was significantly longer in the olarparib group (9.0 months versus 5.4 months; HR 0.44; 95% CI 0.30–0.67; p < 0.0001). 24 Time to second subsequent treatment (TSST) was also longer in the olaparib group (14.9 months versus 9.6 months; HR 0.61; 95% CI 0.42–0.89; p = 0.0111). There was a meaningful clinical delay in TFST and TSST with olaprarib compared with placebo. At 3 years, more than 20% of patients on olaparib had not started subsequent therapy compared with just 3.6% in the placebo group. Time to second disease progression or death (PFS2) was also longer in the olaparib group (16.9 months versus 9.3 months; HR 0.66; 95% CI 0.43–1.02; p = 0.0613). Although not statistically significant, it appears that the benefit of olaparib extends beyond progression of disease as evidenced by almost one-third of patients on olaparib not having second progression of disease at 3 years. 24
Olaparib’s benefit in delaying disease progression led investigators to study its economic impact. The incremental cost–utility ratios were calculated for patients taking maintenance olaparib versus those taking a placebo in the POLO trial. Medical costs included drug acquisition, costs attributed to health states, managing adverse effects, and end-of-life care. All were calculated and considered based on 2018 US dollar values. The study model suggested that maintenance olaparib can potentially be cost-effective in certain scenarios, using a threshold of US$200,000 per quality-adjusted life year (QALY) gained. 25 However, if a threshold of <US$100,000 per additional QALY gained is used, it becomes not cost-effective. The investigators acknowledged that the benefit of overall survival is not conclusive as a major driver of economic evaluation. 25
After the success of the POLO trial, there are several clinical trials currently ongoing with olaparib either by itself or combined with other drugs. NCT04753879 is a phase II trial recruiting untreated metastatic PDAC patients to evaluate the safety and clinical activity of maintenance olaparib and pembrolizumab following multi-agent, low dose chemotherapy with gemcitabine, nab-paclitaxel, capecitabine, cisplatin, and irinotecan (GAX-CI). Another phase II trial is ongoing to study the safety and tolerability of adding pembrolizumab to olaparib, for patients with germline BRCA1/2 mutations who responded to the first line platinum-based therapy in metastatic PDAC [ClinicalTrials.gov identifier: NCT04548752]. Olaparib is also combined with other experimental drugs such as cediranib [AZD2171, vascular endothelial growth factor (VEGF) inhibitor] and AZD6738 (ATR kinase inhibitor) in phase II trials [ClinicalTrials.gov identifiers: NCT02498613, NCT03682289] that are currently recruiting. In an early phase I trial, olaparib and cobimetinib (MEK inhibitor) are being studied in various stages of PDAC to assess the feasibility of collecting tumor tissue for biomarker evaluation prior to and after window therapy with either cobimetinib or olaparib [ClinicalTrials.gov identifier: NCT04005690]. One phase I study is testing combination therapy of olaparib and AZD5153 in patients who progressed through a standard chemotherapy for PDAC and other solid tumors, including lymphomas. This study allows patients’ participation regardless of their BRCA mutational status, but information about their BRCA mutational status should be collected if they were tested for it [ClinicalTrials.gov identifier: NCT03205176]. Other studies of olaparib as a single agent are listed in Table 3 [ClinicalTrials.gov identifiers: NCT01078662, NCT02677038].
Ongoing clinical trials investigating novel targeting agents.

Homologous recombination deficiency.
DNA damage response.
Deleterious BRCA1 or BRCA2 mutations, loss of function mutations in FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN. A known functional mutation in ATM, BACH1 (BRIP1), BARD1, CDK12, CHK1, CHK2, IDH1, IDH2, MRE11A, NBN, PALB2, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD54L.
Last accessed date of clinicaltrials.gov: 8 March 2021.
Other PARP inhibitors in clinical trials
Other than olaparib, there are three PARP inhibitors currently approved for the treatment of different types of cancer. These are rucaparib, niraparib, and talazoparib. A fourth PARP inhibitor, veliparib, received an orphan drug designation for the treatment of advanced squamous non-small cell lung cancer. 26 All PARP inhibitors achieve antitumor activity by trapping PARPs, upregulation of non-homologous end joining activity, and downregulation of homologous recombination repair. 27 Different PARP inhibitors exhibit varying degree of potency in their PARP trapping ability. In vitro studies showed that talazoparib has the highest PARP trapping potency and cytotoxicity. It is 100-fold more potent than olaparib and rucaparib. Compared to olaparib and rucaparib, niraparib also showed superior PARP trapping ability. On the other hand, veliparib is the least potent among the PARP inhibitors. 28
Rucaparib
Rucaparib is currently approved as maintenance therapy for deleterious BRCA mutated (germline and/or somatic) ovarian, fallopian tube, primary peritoneal and castration-resistant prostate cancer. 29 For PDAC, there are three active ongoing clinical trials (Table 3). NCT03140670 is a phase II, single-arm study investigating the safety and effectiveness of rucaparib against advanced pancreatic cancer in patients with deleterious BRCA1/2 or PALB2 mutations. NCT03337087 is a phase I/II trial studying the best dose and side effects of rucaparib and liposomal irinotecan when they are combined with 5-FU and leucovorin in gastrointestinal malignancies including metastatic colorectal, gastroesophageal junction, biliary tract, and pancreas cancers. NCT04171700 is testing rucaparib as treatment for various solid tumors that have deleterious mutations in homologous recombinant repair genes including BRCA1/2. Interestingly, these studies allow participation of patients with not only germline mutations but also somatic mutations. Given that olaparib is only approved for germline BRCA mutations, if any of these studies shows a positive outcome, the indication for PARP inhibitors would expand and it will help patients with somatic mutations (Table 3).
Niraparib
Niraparib is currently approved for first line maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to first line platinum-based chemotherapy. 30 For PDAC, five clinical trials are currently ongoing using niraparib either as a single agent or in combination with other treatment modalities such as immunotherapy and radiation. NIRA-PANC is an open-label, single-arm phase II study looking into the efficacy and safety of niraparib in patients with metastatic pancreatic cancer who progressed through previous lines of chemotherapy. Patients who refuse or cannot tolerate chemotherapy are also eligible. Patients must have germline or somatic mutations in DNA repair genes [ClinicalTrials.gov identifier: NCT03553004]. A phase I study is ongoing to investigate the dose limiting toxicity, maximal tolerable dose, safety, and efficacy of niraparib combined with anlotinib (VEGF receptor inhibitor) in the treatment of advanced solid tumors with homologous recombination repair gene mutations. This trial is being conducted in China and is not available in the United Sates [ClinicalTrials.gov identifier: NCT04764084]. Another phase II study conducted by Dana-Farber Cancer Institute is also testing niraparib as a single agent in the setting of second or further lines of chemotherapy. Patients must harbor either germline deleterious or somatic mutations in BRCA1, BRCA2, PALB2, CHEK2 or ATM genes, to be eligible. The primary endpoint of this trial is PFS, while the primary endpoint of the NIRA-PANC study is the overall response rate (ORR) [ClinicalTrials.gov identifier: NCT03601923]. Both studies are actively recruiting participants as of March 2021. A phase Ib trial is evaluating the effect of niraparib and an experimental monoclonal antibody, TSR-042. The trial will be recruiting patients with germline or somatic BRCA mutated metastatic PDAC that has progressed through standard treatments. Accrual will begin in April 2021 [ClinicalTrials.gov identifier: NCT04673448].
There are three trials investigating niraparib in combination with immunotherapy. NCT04409002 and NCT04493060 are exploring the disease control rate by using an investigational anti-programmed death (PD)-1 agent, dostarlimab, combined with niraparib with or without radiation. Another trial is studying the effectiveness, safety, and anti-tumor activity of niraparib either with ipilimumab (anti-CTLA4 inhibitor) or nivolumab (anti-PD-1). The primary endpoint of this study is PFS. Harboring genetic mutations in DNA repair genes is not mandatory for participation, but the proportion of tumors with homologous recombination deficit and their response to treatment will be analyzed in subgroup analysis as secondary outcome measures [ClinicalTrials.gov identifier: NCT03404960] (Table 3).
Talazoparib
Talazoparib is another PARP inhibitor approved by the US FDA for patients with deleterious or suspected deleterious germline BRCA-mutated, HER2-negative locally advanced or metastatic breast cancer. 31 In PDAC, there is one ongoing clinical trial. NCT04550494 is a basket study evaluating the pharmacodynamic effect of talazoparib for patients with various cancer types that harbor somatic or germline mutations in genes involved in the DNA damage repair process. This trial is currently recruiting participants (Table 3).
Veliparib
Veliparib currently does not have a US FDA approved indication for antitumor treatment. It has the least potent PARP trapping ability compared with other PARP inhibitors, and veliparib failed to improve clinical outcome as a single agent. 32 The US FDA gave veliparib an orphan drug designation for advanced squamous non-small cell lung cancer.26,32 A phase II trial of veliparib as a single agent in patients with previously treated BRCA mutated advanced PDAC did not show any response; however, 25% of the patients had stable disease for more than 4 months. 33 Several early phase trials are evaluating the effects of the combination of veliparib with chemotherapeutic agents. NCT00576654 is a phase I dose escalation trial investigating the best dose and side effects of veliparib when added to irinotecan. NCT01489865 is a phase I/II study to identify the optimal dose of veliparib and its ORR when combined with mFOLFOX6. Veliparib was tolerable at 200 mg twice daily, and the overall response was 26%. In platinum-naive patients with homologous recombination-DDR mutation, there was greater activity with the ORR of 57%. 34 On the other hand, when veliparib was added to cisplatin and gemcitabine in BRCA/PALB2 mutated advanced PDAC patients, it did not show an improved response rate [ClinicalTrials.gov identifier: NCT01585805]. 35 NCT02890355 also failed to show improved overall survival when veliparib is added to mFOLFIRI in biomarker unselected patients. It only showed the increased toxicity of this combined regimen 36 (Table 3).
Resistance to PARP inhibitors
Patients who initially respond to PARP inhibition will unfortunately develop resistance to PARP inhibitors. 37 Several mechanisms for PARP inhibitor resistance have been reported through the in vitro studies of BRCA mutated cell lines from breast and ovarian cancer patients. These include epigenetic regulation of DNA splicing, 38 alternative mRNA spicing, 39 regulation by microRNAs, 40 and restoration of open reading frame to form a nearly full length BRCA by reversion mutations.36,41 These mechanisms lead to restoration of homologous DNA repair or re-establishment of replication fork stability.42,43 Molecular studies of PARP inhibitor resistance have shown that most of these tumors have a hyper-activated ATR/CHK1 pathway. PARP inhibition in BRCA mutated cancer cells causes increased reliance of the ATR/CHK1 pathway for genome stability.42,44 To test this hypothesis of inhibiting PARP and ATR/CHK1 simultaneously would be more efficacious; a phase II trial (NCT03462342) is currently ongoing using olaparib and novel ART inhibitor AZD6738. The trial is currently recruiting participants.
Future implications
Prognosis for PDAC is poor, especially in the metastatic setting. Treatment options are limited. Prior to olaparib, maintenance therapies relied on chemotherapies which typically include fluorouracil or gemcitabine. Chemotherapies are given until the disease no longer responds to the treatment or the patient becomes intolerant to the treatment due to toxicities. Therefore, olaparib is a welcome addition to the armamentarium for the treatment of PDAC. It is currently approved as the maintenance treatment of germline BRCA-mutated PDAC that has not progressed at least 16 weeks of a first-line platinum-based therapy based on impressive PFS data from the POLO trial. At present, the patient population eligible for olaparib maintenance therapy is small due to the relatively low frequency of germline BRCA mutations in PDAC. Outside of germline BRCA mutations, olaparib has also been shown to have activities in other germline and somatic DDR gene mutations such as PALB2, ATM, CDK12. 45 The growing success of PARP inhibitors in this area calls for additional clinical trials to include other DDR gene alterations. Current National Comprehensive Cancer Network (NCCN) guidelines recommend germline testing for all pancreatic cancer patients, using comprehensive gene panels for hereditary cancer mutations, and tumor/somatic gene profiling for patients with locally advanced or metastatic PDAC. Other PARP inhibitors, as monotherapy or in combination with other agents, are being studied in various stages of clinical trials. It is hopeful that the management of PDAC will continuously evolve and eventually lead to better patient outcomes with many ongoing PARP inhibitor trials.
Footnotes
Author contributions
The manuscript has been read and approved for submission by all named authors.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.