
Abstract
Poly(ADP-ribose) polymerase inhibitors (PARPi) are approved as monotherapies in BRCA1/2-mutated (mBRCA1/2) metastatic breast and ovarian cancers, and in advanced pancreatic and metastatic castration-resistant prostate cancers. Differential safety profiles across PARPi necessitate improved mechanistic understanding of inhibitor differences, especially with expansion of PARPi indications and drug combinations. Here, we report in vitro evaluations of PARPi (–/+ PARP trapper temozolomide, TMZ) with reference to total clinical mean concentration average or maximum (tCavg, tCmax), to elucidate contributions of primary pharmacology and structural differences to clinical efficacy and safety. In biochemical assays, rucaparib and niraparib demonstrated off-target secondary pharmacology activities, and in selectivity assays, talazoparib, olaparib, and rucaparib inhibited a broader panel of PARP enzymes. In donor-derived human bone marrow mononuclear cells, only olaparib both increased early apoptosis and decreased the cell viability half inhibitory concentration (IC50) at ≤ tCavg, whereas other PARPi only did so in the presence of TMZ. In cancer cell lines with DNA damage repair mutations, all PARPi decreased cell viability in H1048 but not TK6 cells, and only talazoparib decreased cell growth in DU145 cells at ≤ tCavg concentrations. When combined with low dose TMZ, only talazoparib left-shifted the functional consequences of PARP trapping (S-phase arrest, apoptosis, S-phase double-stranded breaks) and reduced cell viability/growth in TK6 and DU145 cell lines at ≤ tCavg, whereas the other inhibitors required high-dose TMZ. Our study suggests structural differences across PARPi may contribute to differences in PARP selectivity and off-target activities, which along with distinct pharmacokinetic properties, may influence inhibitor-specific toxicities in patients.
Introduction
Approved PARPi Cancer Indications.
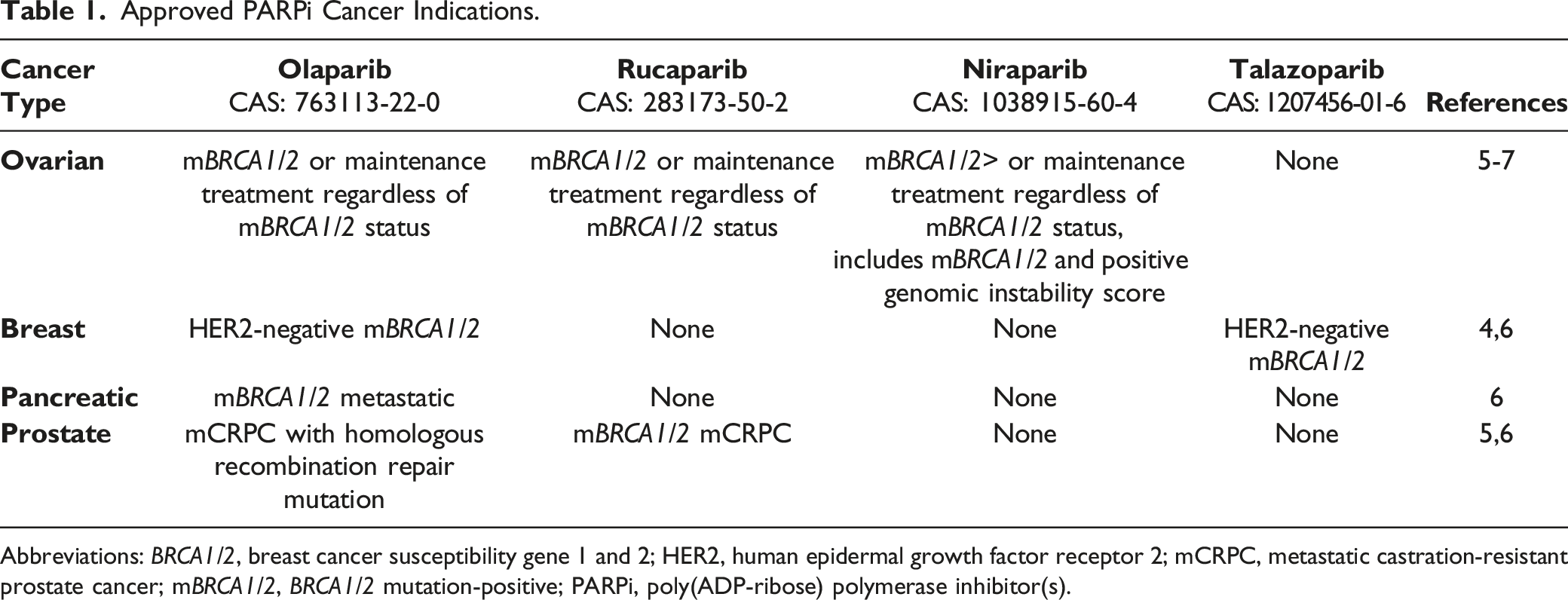
Abbreviations: BRCA1/2, breast cancer susceptibility gene 1 and 2; HER2, human epidermal growth factor receptor 2; mCRPC, metastatic castration-resistant prostate cancer; mBRCA1/2, BRCA1/2 mutation-positive; PARPi, poly(ADP-ribose) polymerase inhibitor(s).
Mechanistically, PARP1 binds to single-strand DNA breaks (SSB) and catalyzes the addition of long poly(ADP-ribose) chains (PARylation), recruits DDR proteins to the DNA lesion, and facilitates chromatin remodeling.12,15,16 The four PARPi demonstrate low nanomolar half inhibitory concentration (IC50) values for PARP enzyme activities, but structural dissimilarities likely contribute to differential PARP trapping potential, referring to the ability of each inhibitor to trap PARP1/2 at sites of damaged DNA.15,17 The functional consequences of PARP trapping are cell cycle arrest, cell growth arrest, double-strand DNA breaks (DSB), replication fork collapse, and cytotoxicity, particularly in cells with mBRCA1/2.17,18 Despite the hypothesis that differential PARP trapping contributes to the cytotoxic potential of each inhibitor, it has been difficult to demonstrate trapping by PARPi alone using in vitro assays and more so using clinically relevant inhibitor concentrations, mostly due to assay detection limitations. Therefore, in vitro assays such as chromatin-bound trapping or single-cell proximity ligation that are used to demonstrate PARPi trapping are conducted in the presence of agents such as methyl methane sulphonate (MMS) or temozolomide (TMZ), which induce SSB lesions and allow for the recruitment of PARP1 and DDR proteins.15,17-19 Even in mBRCA1/2 cell lines, these assays have generally evaluated the inhibitors at concentrations higher than the clinically relevant respective total clinical mean concentration average (tCavg).
Talazoparib has been identified as the most potent PARP trapper, demonstrating the lowest cytotoxicity IC50 values in DDR mutated cancer cell lines in vitro and potent antitumor activity in selected mouse models in vivo.15,18,20 However, the clinical relevance of PARP trapping, as it relates to efficacy of PARPi monotherapy, is unclear. In human epidermal growth factor receptor 2 (HER2)-negative (hormone receptor–positive or triple-negative) mBRCA1/2 metastatic breast cancer the approved doses of talazoparib (1 mg daily) and olaparib (300 mg BID) demonstrate comparable efficacy.4,6 Treatment with up to fourth-line talazoparib has resulted in a median progression-free survival of 8.6 months versus 5.6 months for standard therapy [hazard ratio=0.54; P < 0.001]. Similarly, first-, second-, or third-line olaparib has led to a median progression-free survival of 7.0 months versus 4.2 months for standard therapy [hazard ratio=0.58; P < 0.001]).21-23 In mBRCA1/2 ovarian cancer, olaparib, rucaparib, and niraparib show similar efficacy despite different dosages.5-7,24-26 PARPi combinations with TMZ are currently being explored to evaluate the potential clinical benefit of this combination.18,27
PARPi-related hematologic dose-limiting toxicities (DLTs) can be generally managed through dose interruptions and reductions, although the cell types affected differ for each inhibitor.4-7,28-30 Nonhematologic toxicities associated with PARPi can also have a substantial impact on tolerability and play an additional critical role in determining the optimal therapeutic index, particularly when exploring PARPi combinations.4-7,29,30 Furthermore, the underlying mechanisms behind the slightly differential clinical safety profiles among PARPi have not been fully elucidated. To understand the implications of structural differences and the trapping potential of PARPi, and to establish potential correlations with the reported clinical safety profiles, we compared the inhibitors in biochemical assays for PARP selectivity and for off-target secondary pharmacology activities. To understand the functional consequences of PARP trapping, PARPi were also evaluated in the absence and presence of TMZ (–/+ TMZ) for cytotoxicity and early apoptosis using healthy donor-derived human bone marrow mononuclear cells (hBMMNC) and in endpoint-based cellular assays (cytotoxicity/cell growth, apoptosis, cell cycle distribution, and DSB) using cancer cell lines with identified DDR mutations (SCLC, lymphoblastoid, and mCRPC). All endpoint evaluations were conducted with reference to tCavg or total clinical concentration maximum (tCmax) to ensure meaningful clinical relevance.
Materials and Methods
PARP Selectivity, Secondary Pharmacology, and Kinase Selectivity
The four PARPi were evaluated for selectivity in a 12-PARP panel biochemical enzymatic assay, while off-target secondary pharmacology activity and kinase selectivity were evaluated against a broad panel of 85 receptors, enzymes, and ion channels (see Supplementary Methods).
Cell Line Culture
The SCLC cell line (NCI-H1048) purchased from American Type Culture Collection (ATCC; Gaithersburg, MD) was genotyped (mTP53; ATR, S1693G; HDAC2, K105fs) and cultured in DMEM-F12 medium (Gibco BRL, Gaithersburg, MD) supplemented with 0.005 mg/mL insulin, 0.01 mg/mL transferrin, 30 nM sodium selenite, 10 nM hydrocortisone, 10 nM beta-estradiol, 2 mM L-glutamine, and 5% FBS in a 37 °C, 5% CO2, and 98% humidity incubator. The cells were plated (10,000 cells/mL) in 384-well white-wall clear-bottom plates (Fisher Scientific, Waltham, MA) for 24 hours before treatment with test compounds. TK6 (ATCC CRL-8015) human lymphoblast cells and DU145 (ATCC HTB-81) human metastatic prostate carcinoma cells were purchased from the ATCC (Gaithersburg, MD) and genotyped ([TK6: MGMT negative; mMLH1], [DU145: BRCA1, E962K; BRCA2, S2284L; mFANCl; MLH1, A586V]), and frozen stocks were maintained in liquid nitrogen. Working cell stocks were maintained in log phase for 2 to 15 passages in RPMI 1640 plus L-glutamine, FBS, penicillin-streptomycin, and sodium pyruvate (Complete Culture Medium [CCM]; Gibco BRL, Gaithersburg, MD) in a 37 °C, 5% CO2, and 98% humidity incubator. Stock cultures were further maintained in log phase (4 x 105 and 1 x 106 cells/mL) during experimental set ups and plated in 96-well plates in CCM at 5.5–6.5 x 104 cells/mL (∼6000 cells per well) for 48 hours before test article exposure. Mycoplasma testing was routinely performed on TK6 cells (last test November 2020), whereas H1048 and DU145 cells were utilized within a year after receipt and no additional mycoplasma testing was performed beyond the certification received with the purchase.
Human Bone Marrow Culture
Healthy donor hBMMNCs (mixture of hematopoietic stem and progenitor cells) were obtained from Lonza (Walkersville, MD) and cultured in Stemline® Hematopoietic Stem Cell Expansion Medium (HSCM; Sigma, St. Louis, MO), which was supplemented with 10% FBS and cytokines (R&D systems, Minneapolis, MN) including stem cell factor (25 ng/mL), erythropoietin (3 U/mL), granulocyte colony-stimulating factor (10 ng/mL), granulocyte-macrophage colony-stimulating factor (10 ng/mL), thrombopoietin (15 ng/mL), interleukin (IL)3 and IL6 (10 ng/mL), and Fms-related receptor tyrosine kinase 3 (Flt3) ligand (25 ng/mL). The cells were incubated in a 37 °C, 5% CO2, and 98% humidity incubator. The hBMMNCs were plated at 5 x 105 cells/mL in 96-well white-wall clear-bottom plates (Fisher Scientific, Waltham, MA) in triplicate for each test article concentration and treated at 24 hours post-plating.
Compound Treatment
The cellular assays were conducted across a range of concentrations encompassing the pharmacologically relevant clinical tCavg for each inhibitor. Similarly, the two TMZ concentrations used in the cellular assays represent clinically relevant exposures corresponding to a low and a full approved dose of TMZ. Compound assessment included the four PARPi, talazoparib (PF-06944076, Albany Molecular Research, Inc., Albany, NY), olaparib (Pfizer Inc.), rucaparib (APExBIO Cat# A4156, Batch 1), and niraparib (Pfizer Inc.), as well as the PARP trapping alkylating agent TMZ (Sigma, Cat# T2577 Lots: 00001788 [DU145 experiments] and 046M4753V [TK6 experiments], San Diego, CA).
Preparation of Compound Concentrations
For hBMMNC, H1048, TK6, and DU145 cells, a 200x unique stock of each inhibitor was prepared in DMSO, the concentration of which was guided by the mean tCavg. For the hBMMNC and H1048 cells, 9-serial dilutions were made from each stock, generating 10 sub-stock concentrations for each inhibitor. Two μL of each sub-stock or vehicle was added to 200 μL of complete media in the 96-well plates containing hBMMNC or H1048 cells and incubated for 5 days. For each experimental setup, four 96-well cell plates were prepared. Plate 1 contained vehicle and positive controls, Plate 2 contained PARPi without TMZ, and Plates 3 and 4 contained PARPi with the low (30 μM) and high (100 μM) dose of TMZ, respectively. For TK6 and DU145 cells, 24-serial dilutions were made from each stock, generating 24 sub-stock concentrations for each inhibitor. Three μL of each sub-stock or vehicle was added to 600 μL of complete media in the 96-well plates containing TK6 or DU145 cells and incubated for approximately 4 hours and 24 hours, respectively. For each experimental setup, a total of four 96-well plates were prepared. Plate 1 contained vehicle and positive controls, Plate 2 contained PARPi without TMZ, and Plates 3 and 4 contained PARPi with the low (43 μM) and high (128 μM) dose of TMZ, respectively. For each cell type, the final concentrations of the test article in the media ranged from 2.4 x 10-6 to 20 μM for talazoparib, 1.8 x 10-5 to 150 μM for olaparib, 3.0 x 10-5 to 250 μM for rucaparib, and 2.4 x 10-5 to 200 μM for niraparib. Therefore, the mean tCavg (nM) exposures for talazoparib (15.7 nM), olaparib (4700 nM), rucaparib (2534 nM), and niraparib (1608 nM) were included in the tested final concentration range and the TMZ doses were selected to represent the mean clinical tCavg exposures at low (37.5 mg/m2) and high (150 mg/m2) clinical doses of TMZ.
Cell Viability and Early Apoptosis Assays in hBMMNC and H1048 Cells
Cell viability was assessed on Day 5 following 4 days of exposure to vehicle or compounds (PARPi –/+ TMZ), by measuring intracellular ATP content using bioluminescence readout per the manufacturer’s recommended protocol (CellTiter-Glo® luminescent cell viability assay kit; Promega, Madison, WI) and the Safire2 microplate reader (TECAN, Switzerland). Early apoptosis was assessed following compound treatment for 24 hours, using bioluminescence readout per the manufacturer’s protocol for the Caspase-Glo® 3/7 activation assay (Promega, Madison, WI) and the Safire2 microplate reader (TECAN, Switzerland). Solubilized caspase 3/7 substrate (25 μL) was added to each well containing 50 μL of cell suspension and incubated at room temperature for 30 minutes before measuring the bioluminescence. The dose-response relationship and statistical significance were analyzed using GraphPad Prism software version 8.
Cell Viability and Single Cell DDR Assay in TK6 and DU145 Cell Lines
For both the TK6 and DU145 cell lines, cell viability was assessed using the propidium iodide (PI) exclusion method and fluorescence-activated cell sorting (FACS) analysis. The single-well DDR assay for both cell lines included assessment of phosphorylated H2A.X variant histone (γH2AX) and cleaved caspase-3 (CC-3) levels in a cell cycle (PI staining)-dependent manner. An increase in the level of γH2AX (phosphorylation at serine 139 detected using anti-γH2AX antibody) is associated with induction of DSB and is a marker for the recruitment of DDR proteins to the site of the damaged DNA, 31 whereas CC-3 is an early marker of commitment of viable cells to apoptosis. 32 Since early apoptosis-initiated DNA fragmentation can also increase γH2AX levels, 33 percent of CC-3-negative/γH2AX+ cells reflected DSB that were directly compound-induced. Before preparation for flow cytometry harvest, DU145 were lifted from culture wells and made into cell suspensions using 0.25% trypsin EDTA (Gibco BRL, Gaithersburg, MD). Following 4- or 24-hour treatment with the test compounds, the 96-well plates were washed with PBS, fixed with 0.25% formaldehyde/PBS solution, permeabilized using ice-cold 100% methanol, and stored at −20 °C. For γH2AX and CC-3 detection in a cell cycle-dependent manner, the samples were removed from the freezer and centrifuged at 220 x g for 6 minutes; the methanol was aspirated from the plates using a Biotek ELX plate washer, and samples were resuspended in ∼200 µL of 0.1% Tween 80/PBS permeabilizing solution (Sigma; Catalog #14780-100 mL; CAS # 9005-65-6) for 15 minutes at room temperature. The plates were recentrifuged, the supernatant aspirated, and the cells stained with 100 µL of antibody cocktail containing Alexa Fluor 488-labeled anti-CC-3 antibody (1:50, Cell Signalling Technology Cat# 9603, RRID:AB_11179205) and Alexa Fluor 647-labeled anti-phospho-histone H2AX(ser139) (1:50, BD Biosciences Cat# 560447, RRID:AB_1645414) for 1 hour at 37 °C. Following incubation with the antibodies, the cells were washed with cold PBS, and DNA was counterstained with PI (25 µg/mL, Molecular Probes Catalog #3566) containing RNase A (0.2 mg/mL, Macherey-Nagel Catalog # 740505) for the cell cycle analyses. The details of the FACS analyses are summarized in the Supplemental Methods.
Statistical Analyses
The dose-response analysis for the single cell DDR assay endpoints for the TK6 and DU145 cell lines were completed using ToxCast pipeline, an R package designed to process high-throughput screening data. 34 The dose-response analysis for the hBMMNC and H1048 cells were conducted using GraphPad Prism. Summary figures of inhibitor selectivity and cellular assay data were produced using Spotfire. The details of the statistical analysis are summarized in the Supplemental Methods.
Availability of Data and Material
Upon request and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Results
Summary of PARP Selectivity and Secondary Pharmacology Assays
The IC50 values for talazoparib, olaparib, rucaparib, and niraparib, as well as the vendor-provided olaparib control were determined using dose-response curves for the 12 PARP enzymes in the selectivity panel and are summarized in Figure 1A (actual values in Supplementary Table 1). At the clinically approved doses, the tCavg exposure values for talazoparib, olaparib, rucaparib, and niraparib are 15.7, 4700, 2534, and 1608 nM, respectively. At ≤ tCavg concentrations, niraparib appears to be the most selective PARP1/2 inhibitor, whereas talazoparib, olaparib, and rucaparib are expected to inhibit additional PARP enzymes. At clinically efficacious exposures, inhibition is expected with talazoparib and rucaparib for PARP3 and both tankyrase enzymes (TNKS1 [PARP5a] and TNKS2 [PARP5b]) and with rucaparib also for PARP10. The PARP selectivity profile of each inhibitor likely contributes to some of the safety endpoints as seen in patients. Summary of (A) PARP selective activity and (B) secondary polypharmacology activity. (A) PARP selectivity across the four inhibitors displayed as points for each PARP enzyme based on the respective IC50 values (nM) and sized based on the log10 exposure ratio or log10(IC50/Cavg). The empty circles designate inactive or negative results up to 10,000 nM. IC50 values (nM) for each inhibitor were determined using dose-response curves for the respective PARP enzyme assay. The solid and dotted lines represent the tCavg and tCmax exposure for each inhibitor. Experiments were performed in triplicate. (B) Additional off-target profiles were generated for rucaparib and niraparib since there was target inhibition or binding greater than 50% at the initial tested concentrations of 10 µM. Ki (empty circle) and IC50 (filled circle) potency values (nM) are displayed relative to their respective total Cavg and Cmax values and sized accordingly based on their log10 exposure ratio or log10 (Potency/Cmax for the ion channels/receptors and Potency/Cavg for the kinase targets). The solid and dotted lines represent the tCavg and tCmax exposure for each inhibitor. Experiments were performed in triplicate. Abbreviations: 5-HT4, 5-hydroxytryptamine receptor 4 (non-selective sigma receptor; HTR4); CDK, cyclin-dependent kinase; DAT, dopamine transporter; Ki, inhibitory constant; IC50 half maximal inhibitory concentration; MAO, monoamine oxidase; NET, norepinephrine transporter; PARP, poly(ADP-ribose) polymerase; PASK, PAS domain containing serine/threonine kinase; PIM3, Pim-3 proto-oncogene, serine/threonine kinase; SERT, serotonin transporter; tCavg, total concentration average; tCmax, total concentration maximum.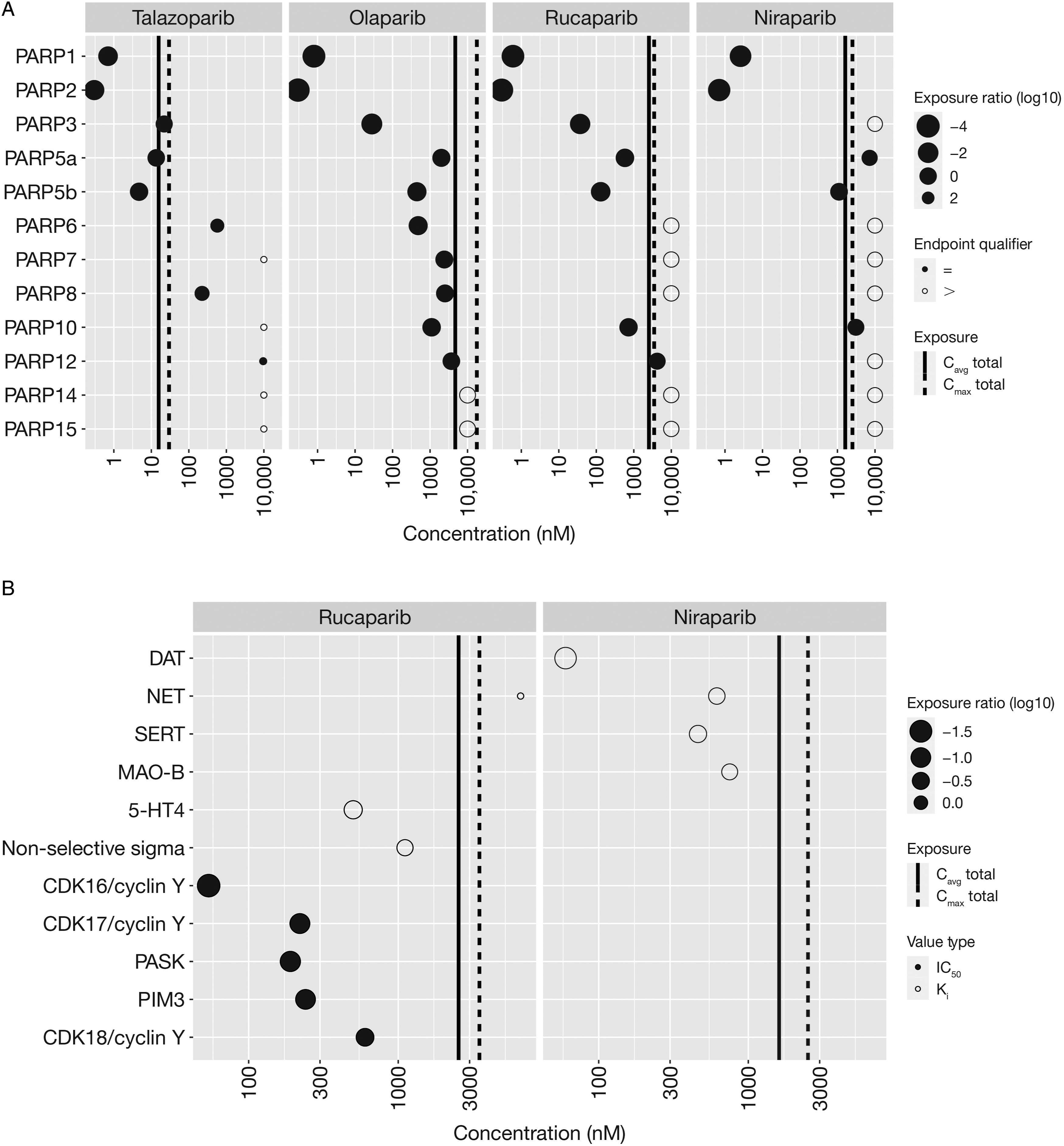
In the off-target secondary pharmacology evaluation, summarized in Figure 1B (actual values in Supplementary Table 2), talazoparib and olaparib at ≤ tCmax concentrations showed ≤30% activity for the 85 evaluated targets, indicating minimal potential for off-target liability. At ≤ tCmax exposure concentrations, rucaparib and niraparib showed ≥50% target inhibition for 13/85 and 17/85 targets, respectively. Specifically, rucaparib inhibited binding at the non-selective sigma receptor (5-HT4 [5-hydroxytryptamine receptor 4; non-selective sigma receptor; HTR4]) and several kinase enzyme activities, whereas niraparib inhibited binding at the dopamine (DAT), norepinephrine (NET), and serotonin (SERT) reuptake transporters (Triple Reuptake Inhibitor), and at monoamine oxidase (MAO)-B. The kinases inhibited by rucaparib at ≤ tCmax and tCavg concentrations include CDK16, 17, and 18/cyclin Y, and the serine/threonine kinases Per-Arnt S domain-containing kinase (PASK) and Pim-3 proto-oncogene, serine/threonine (PIM3).
Summary of Selected Adverse Events in ≥10% Patients in Maintenance Treatment of Recurrent Germline mBRCA1/2 Ovarian Cancer and Correlation With PARP Selectivity and Secondary Off-Target Activities.
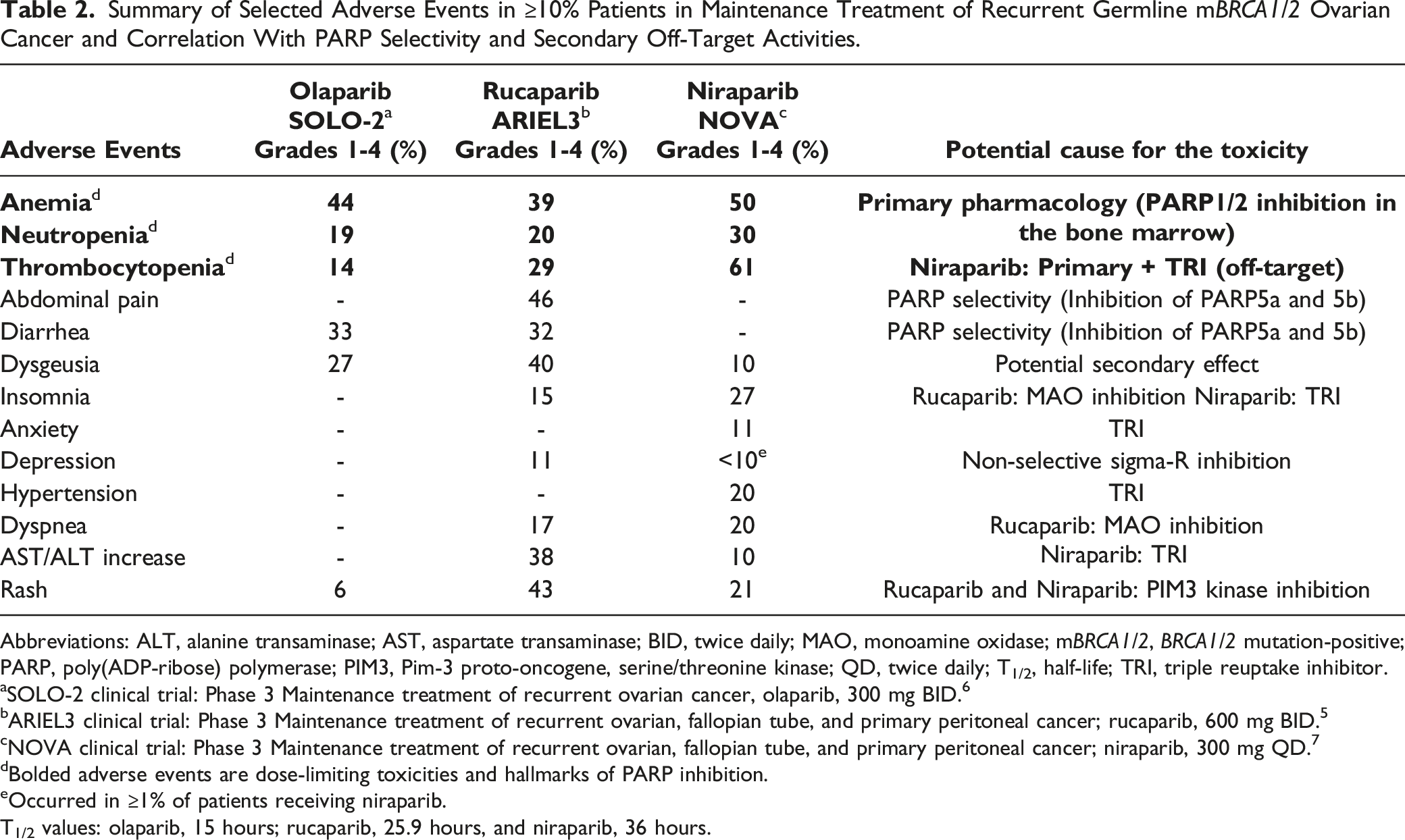
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; BID, twice daily; MAO, monoamine oxidase; mBRCA1/2, BRCA1/2 mutation-positive; PARP, poly(ADP-ribose) polymerase; PIM3, Pim-3 proto-oncogene, serine/threonine kinase; QD, twice daily; T1/2, half-life; TRI, triple reuptake inhibitor.
aSOLO-2 clinical trial: Phase 3 Maintenance treatment of recurrent ovarian cancer, olaparib, 300 mg BID. 6
bARIEL3 clinical trial: Phase 3 Maintenance treatment of recurrent ovarian, fallopian tube, and primary peritoneal cancer; rucaparib, 600 mg BID. 5
cNOVA clinical trial: Phase 3 Maintenance treatment of recurrent ovarian, fallopian tube, and primary peritoneal cancer; niraparib, 300 mg QD. 7
dBolded adverse events are dose-limiting toxicities and hallmarks of PARP inhibition.
eOccurred in ≥1% of patients receiving niraparib. T1/2 values: olaparib, 15 hours; rucaparib, 25.9 hours, and niraparib, 36 hours.
Summary of Selected Adverse Events in ≥10% of Patients With Germline mBRCA1/2 HER2-Negative Metastatic Breast Cancer and Correlation With PARP Selectivity And Secondary Off-Target Activities.
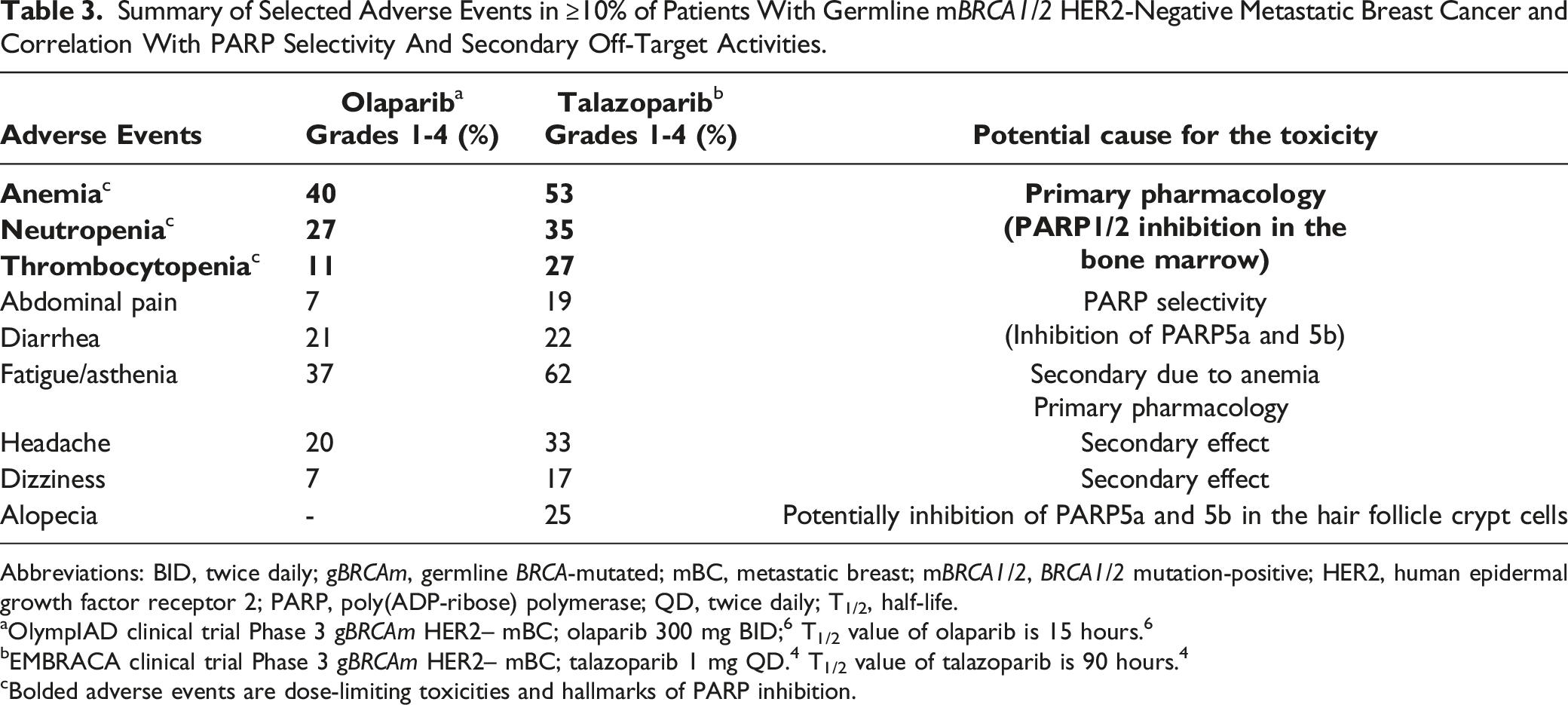
Abbreviations: BID, twice daily; gBRCAm, germline BRCA-mutated; mBC, metastatic breast; mBRCA1/2, BRCA1/2 mutation-positive; HER2, human epidermal growth factor receptor 2; PARP, poly(ADP-ribose) polymerase; QD, twice daily; T1/2, half-life.
aOlympIAD clinical trial Phase 3 gBRCAm HER2– mBC; olaparib 300 mg BID; 6 T1/2 value of olaparib is 15 hours. 6
bEMBRACA clinical trial Phase 3 gBRCAm HER2– mBC; talazoparib 1 mg QD. 4 T1/2 value of talazoparib is 90 hours. 4
cBolded adverse events are dose-limiting toxicities and hallmarks of PARP inhibition.
Safety of PARPi (–/+ TMZ) in hBMMNCs
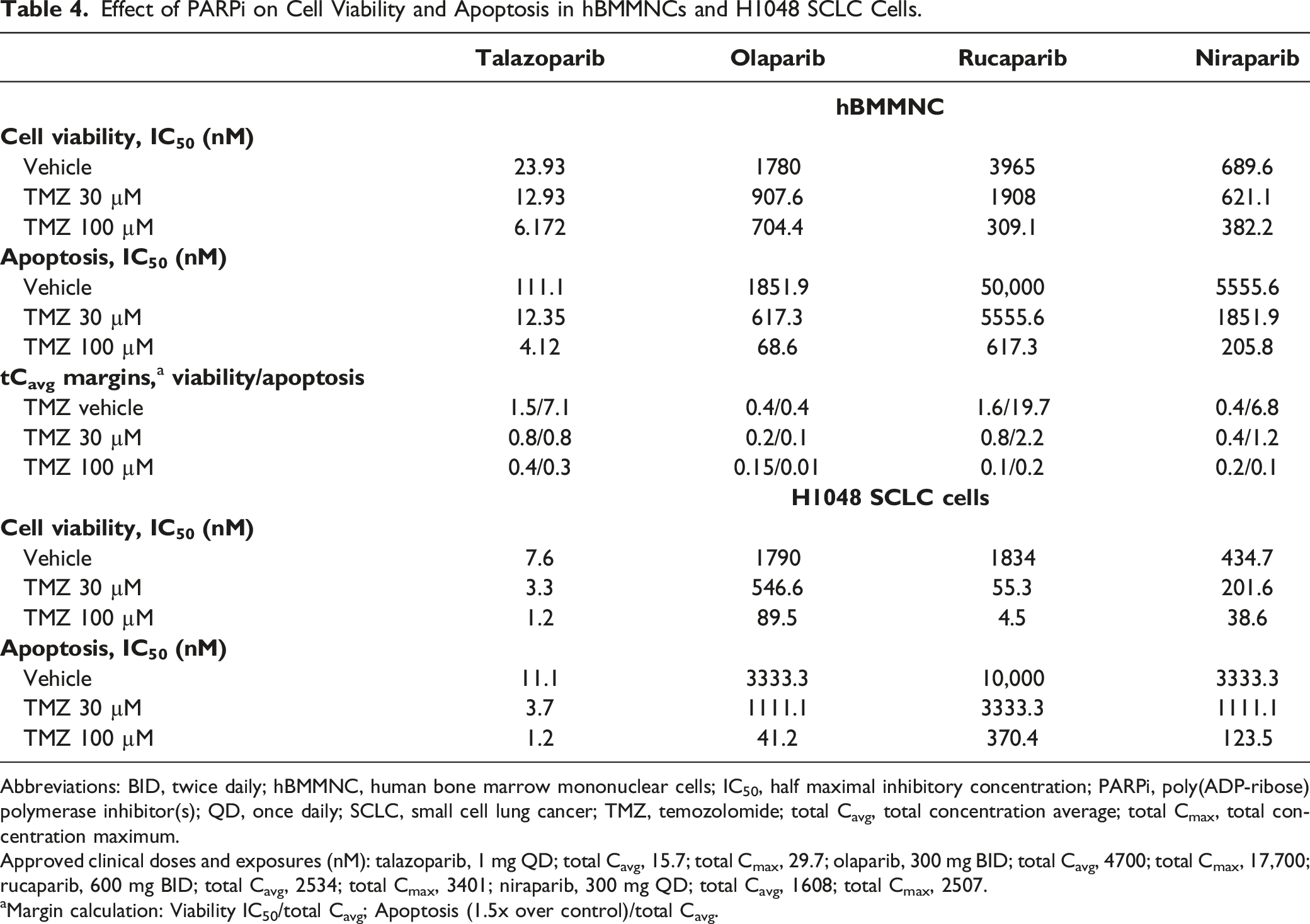
To evaluate the direct effect on hematologic toxicity, the PARPi (–/+ TMZ) were tested in healthy donor hBMMNC (no DDR mutations) for effects on cell viability (4-day treatment) and early apoptosis (24-hour treatment). The IC50 values for each endpoint are summarized in Figure 2, the actual values along with the respective safety margins (IC50 values/clinical tCavg) are summarized in Table 4. In the absence of TMZ, the cell viability IC50 values were lower than tCavg for olaparib (1780 nM) and niraparib (690 nM), resulting in low safety margins (0.4x each) but higher than tCavg for talazoparib (23.9 nM) and rucaparib (3965 nM), resulting in better safety margins (1.5x and 1.6x, respectively). TMZ caused dose-dependent synergistic left-shifted IC50 values, which decreased cell viability safety margins for all four inhibitors (Table 4). To evaluate the effect on early apoptosis, the CC-3 level increases (1.5x compared with vehicle control) were measured and safety margins calculated. In absence of TMZ, only olaparib increased CC-3 levels at lower than tCavg concentration, and the safety margins were comparable to that of cell viability (0.4x). Talazoparib, rucaparib, and niraparib did not cause an increase in early apoptosis at the tCavg concentrations, resulting in higher safety margins for early apoptosis (7.1x, 19.7x, and 6.8x, respectively). TMZ, which is expected to increase PARP trapping, dose-dependently left-shifted the early apoptosis safety margins, resulting in cell viability safety margins similar to the high TMZ dose for all PARPi (Table 4). TMZ alone did not affect early apoptosis. Summary of cellular assay data in hBMMNC, H1048 SCLC, TK6 lymphoblastoid, and DU145 mCRPC cells. Potency estimates (IC50 values in nM) for the four inhibitors across hBMMNC and the four cancer cell lines are displayed as points for cell viability/growth and apoptosis (all five cell types) and for select markers of DNA damage response (TK6 and DU145). The data point represents the concentration at which there was a 50% decrease in cell viability or growth compared with vehicle control. Apoptosis values represent the concentration at which there was 1.5x increase compared with vehicle control. S-phase accumulation values represent the concentration at which there was 2x accumulation compared with vehicle. S-phase γH2AX represents ACC values generated for DSB. The points are sized based on their log10 exposure ratio or log10 (IC50/Cavg). TMZ co-exposures are color coded as no TMZ (black), low concentration of TMZ (light blue) and high concentration of TMZ (dark blue). Empty circles denote inactive tests up to the maximal tested concentration and denoted with the greater than symbol. The solid lines represent the tCavg exposure for each inhibitor. hBMMNC and H1048 SCLC cell lines were tested with TMZ concentrations of 30 and 100 μM, whereas the TK6 lymphoblastoid and DU145 androgen receptor inhibitor–resistant mCRPC cell lines were tested with TMZ concentrations of 43 and 128 μM. IC50 values for cell viabilities were determined for hBMMNCs and H1048 cells using dose response curves. For TK6 (cell viability based on PI exclusion) and DU145 (cell growth based on population doubling), the reported concentrations represent 50% decrease compared with vehicle. Experiments were performed in triplicate. Abbreviations: γH2AX, phosphorylated H2A.X variant histone; ACC, activity concentration at cut-off; DSB, double-strand DNA breaks; hBMMNC, human bone marrow mononuclear cell; IC50, half maximal inhibitory concentration; mCRPC, metastatic castration-resistant prostate cancer; PI, propidium iodide; SCLC, small cell lung cancer; tCavg, total concentration average; tCmax, total concentration maximum; TMZ, temozolomide. Effect of PARPi on Cell Viability and Apoptosis in hBMMNCs and H1048 SCLC Cells. Abbreviations: BID, twice daily; hBMMNC, human bone marrow mononuclear cells; IC50, half maximal inhibitory concentration; PARPi, poly(ADP-ribose) polymerase inhibitor(s); QD, once daily; SCLC, small cell lung cancer; TMZ, temozolomide; total Cavg, total concentration average; total Cmax, total concentration maximum. Approved clinical doses and exposures (nM): talazoparib, 1 mg QD; total Cavg, 15.7; total Cmax, 29.7; olaparib, 300 mg BID; total Cavg, 4700; total Cmax, 17,700; rucaparib, 600 mg BID; total Cavg, 2534; total Cmax, 3401; niraparib, 300 mg QD; total Cavg, 1608; total Cmax, 2507. aMargin calculation: Viability IC50/total Cavg; Apoptosis (1.5x over control)/total Cavg.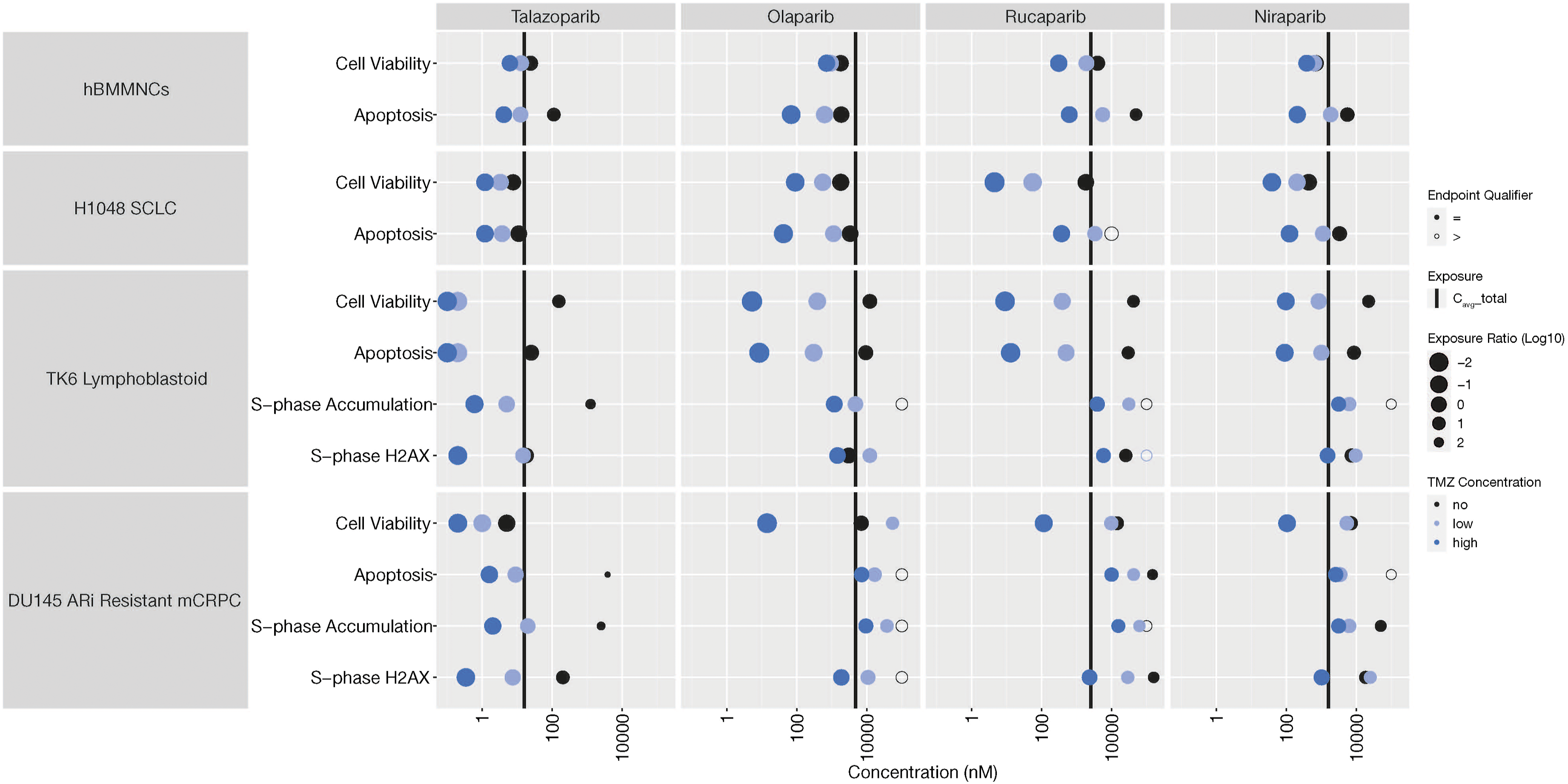
Efficacy of PARPi (–/+ TMZ) in H1048 SCLC Cell Lines
The PARPi (–/+ TMZ) were compared in cell viability and early apoptosis assays in H1048 cells (mTP53; ATR, S1693G; HDAC2, K105fs) at concentrations similar to those used for hBMMNC. Summary of the IC50 values from the 4-day cell viability assays and 24-hour early apoptosis for each inhibitor (–/+ TMZ) are summarized in Figure 2, the values are tabulated in Table 4. In the absence of TMZ, all the cell viability IC50 values (talazoparib [7.6 nM], olaparib [1790 nM], rucaparib [1834 nM], and niraparib [435 nM]) were lower than the respective tCavg exposures, demonstrating that H1048 cells are sensitive to all PARPi at clinically relevant exposures. In presence of TMZ, there was a synergistic left-shift in the IC50 values for each inhibitor (Table 4), although TMZ alone (30 or 100 µM) did not impact cell viability (Figure 2). To understand the effect of early apoptosis on cell viability, the increase in CC-3 levels (1.5x compared with vehicle control) were evaluated following 24-hour treatment. In the absence of TMZ, CC-3 levels increased at < tCavg concentrations with talazoparib and olaparib but at > tCavg concentrations with rucaparib and niraparib. In the presence of TMZ, CC-3 increases were left shifted at less than the respective tCavg concentrations for all four inhibitors. TMZ alone had no effect on early apoptosis or cell viability but dose-dependently potentiated the efficacy of the PARPi in H1048 cells.
Efficacy of PARPi (–/+ TMZ) in TK6 Lymphoblastoid Cell Lines
Dose Response Analysis in TK6 and DU145 (–/+ TMZ) at 24 Hours Post Treatment.
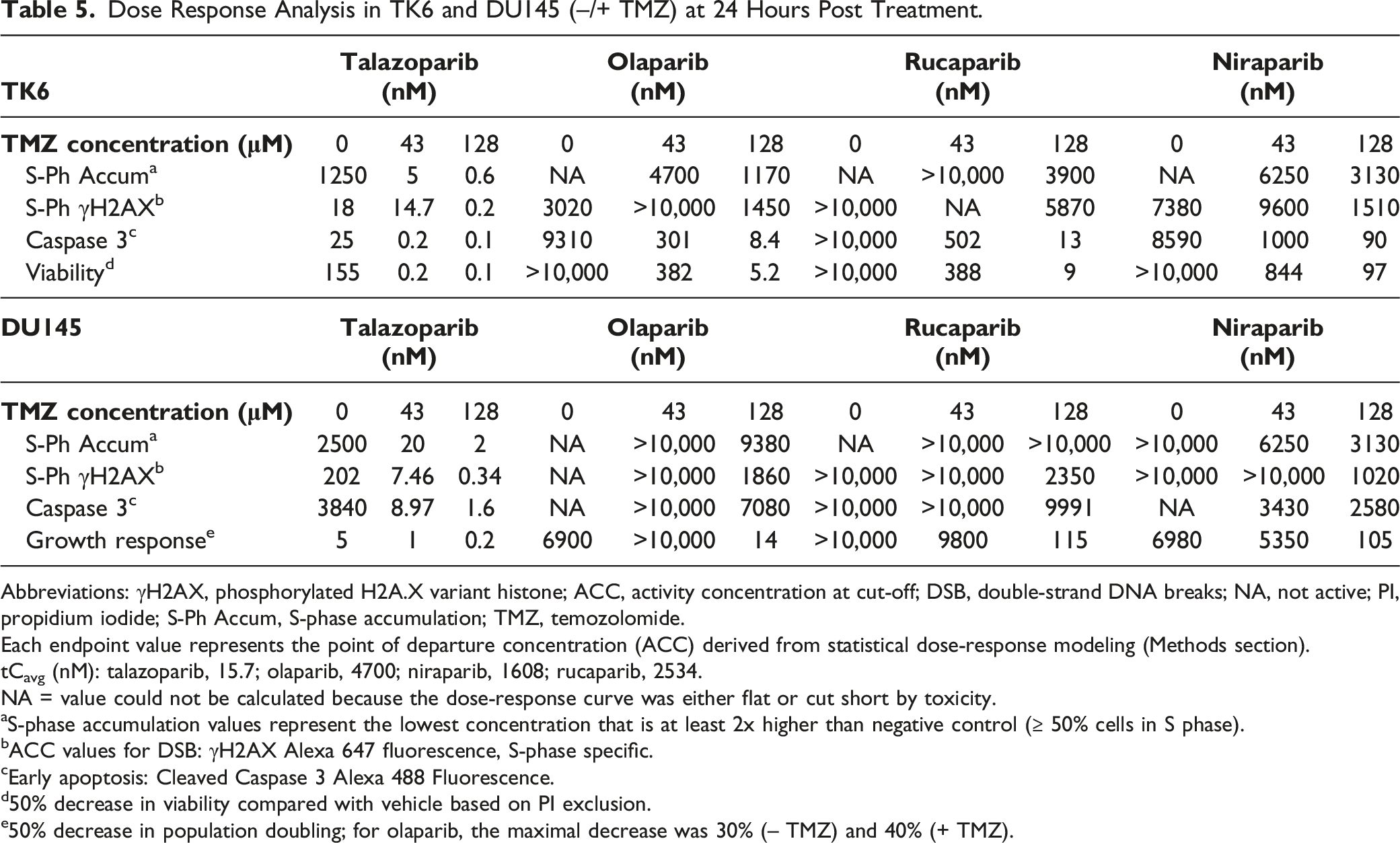
Abbreviations: γH2AX, phosphorylated H2A.X variant histone; ACC, activity concentration at cut-off; DSB, double-strand DNA breaks; NA, not active; PI, propidium iodide; S-Ph Accum, S-phase accumulation; TMZ, temozolomide.
Each endpoint value represents the point of departure concentration (ACC) derived from statistical dose-response modeling (Methods section).
tCavg (nM): talazoparib, 15.7; olaparib, 4700; niraparib, 1608; rucaparib, 2534.
NA = value could not be calculated because the dose-response curve was either flat or cut short by toxicity.
aS-phase accumulation values represent the lowest concentration that is at least 2x higher than negative control (≥ 50% cells in S phase).
bACC values for DSB: γH2AX Alexa 647 fluorescence, S-phase specific.
cEarly apoptosis: Cleaved Caspase 3 Alexa 488 Fluorescence.
d50% decrease in viability compared with vehicle based on PI exclusion.
e50% decrease in population doubling; for olaparib, the maximal decrease was 30% (– TMZ) and 40% (+ TMZ).
In absence of TMZ, the increases in CC-3+ early apoptosis ACC values were observed only at PARPi concentrations > tCavg for each inhibitor and therefore were not clinically meaningful. TMZ dose-dependently resulted in synergistic increases in CC-3+ positive cells and left-shifted ACC values at PARPi concentrations that were significantly lower than the respective tCavg for all inhibitors. The ACC values for S-phase-specific γH2AX+ MFI at 24 hours following treatment with PARPi alone were similar to the tCavg for talazoparib (18 nM) and olaparib (3020 nM) but > tCavg for niraparib (7380 nM) and rucaparib (25,500 nM). Only with high-dose TMZ that resulted in sufficient PARP trapping for rucaparib and niraparib, the S-phase-specific γH2AX+ ACC values were < tCavg exposures (Table 5). TMZ alone also caused an approximately 2-fold increase in γH2AX levels compared with the vehicle control. In the absence of TMZ, decreases in cell viability (50% decrease versus vehicle) by PARPi were observed at higher concentrations than the respective tCavg. Similar to the effects on early apoptosis, TMZ also dose-dependently resulted in a synergistic decrease in PARPi-induced cell viability at concentrations lower than the respective tCavg (Table 5). The TK6 cells appear to be less sensitive to PARPi-induced effects on cell viability compared with H1048 cells. Additional PARP trapping by TMZ was required to affect multiple DDR parameters although talazoparib and olaparib required the low dose whereas rucaparib and niraparib required the higher dose of TMZ.
Efficacy of PARPi (–/+ TMZ) in DU145 mCRPC Cell Lines
DU145 are hormone insensitive, androgen receptor–negative cells (mutations: BRCA1, E962K; BRCA2, S2284L; mFANCl; MLH1, A586V) that have a cell doubling time of about 20 hours. Therefore, the acute effects of the PARPi (–/+ TMZ) at 24 hours post-treatment were assessed through stepwise evaluation of cell cycle distribution (% cell each phase), early apoptosis (1.5x increase over vehicle control), S-phase specific DSB (ACC values), and cell growth arrest that represent the hallmarks of PARPi activity. The data are summarized in Figure 2, the values are tabulated in Table 5, and additional profiles are summarized in Supplementary Figures 3 and 4. The distribution of cells in the vehicle group at 24 hours post treatment was 65% in G1, 20% in S, and 15% in G2/M (data not shown) and the concentration at which the S-phase cells increased by 2-fold (compared with vehicle control) are tabulated for each inhibitor in Table 5. In absence of TMZ, the PARPi did not induce meaningful S-phase accumulation. In the presence of low-dose TMZ, PARP trapping was sufficient to reduce the concentration for S-phase accumulation slightly above the tCavg (20 nM) for talazoparib alone. PARP trapping was sufficient at high-dose TMZ to cause S-phase accumulation at ≤ tCavg concentrations for all four inhibitors.
In the absence of TMZ, PARPi did not increase CC-3+ early apoptosis in DU145 cells. In the presence of low dose TMZ, only talazoparib resulted in a synergistic increase (1.5x over vehicle control) in CC-3+ cells at < tCavg (9 nM), and the trend continued (1.6 nM) with high-dose TMZ (Table 5). For olaparib, rucaparib, and niraparib, even with high-dose TMZ the increases in CC-3+ cells were at non-meaningful concentrations. In the absence of TMZ, the S-phase-specific γH2AX+ ACC values were higher than the tCavg exposure for all PARPi. In presence of low dose TMZ, and similar to accumulation of cells in S-phase, the S-phase-specific γH2AX+ ACC values were drastically left-shifted for talazoparib and the trend continued with high-dose TMZ. For the other PARPi the S-phase-specific γH2AX+ ACC values were lower than or closer to the tCavg concentrations, only with high-dose TMZ whereas TMZ alone did not affect γH2AX+ levels. Cell growth arrest (50% decrease in cell growth compared with vehicle) in DU145 cells was observed at significantly lower (5 nM) concentrations than the tCavg with talazoparib alone (– TMZ) and the effect was further left-shifted in the presence of low and high-dose TMZ to 1 and 0.2 nM, respectively (Table 5). Only with high-dose TMZ did the other three inhibitors decrease cell growth (40% maximum decrease for olaparib) at ≤ tCavg concentration. Therefore, decrease in the DU145 cell growth is selectively and meaningfully responsive to talazoparib alone.
Discussion
It is widely accepted that clinical efficacy and hematologic toxicities observed with PARPi result from pharmacological effects on cancer cells and the bone marrow, due to PARP1/2 enzyme inhibition and increased PARP trapping.19,28-30 In recent years, it has been suggested that the PARP trapping function of PARPi contributes to the exaggerated hematologic toxicity of PARPi and studies evaluating the adverse event profile of talazoparib, which is considered a potent PARP trapper, are often used to support such conclusions.15,19,35 However, most in vitro studies have used the inhibitors in the presence of exogenous PARP trappers such as TMZ and MMS at higher than clinically relevant concentrations to reach such conclusions.15,19,35 In this study we focused on correlating the differential clinical toxicity profiles of the inhibitors with experimental readouts conducted at clinically relevant concentrations. Using clinically relevant low and high exposures, we have also used TMZ, which is currently in several PARPi combination clinical trials (NCT02446704, NCT01085422, and NCT03672773), to demonstrate how increased trapping might modulate PARPi-induced safety and efficacy at clinically relevant PARPi and TMZ concentrations.
Our study suggests that the structural differences of PARPi likely contribute to some of the inhibitor-specific hematologic and also nonhematologic toxicities in patients, due to differences in PARP selectivity and off-target secondary pharmacology activities at ≤ tCavg and/or tCmax exposures. Additionally, other reports highlight that distinct pharmacokinetic properties may play a role.4-7,19 For example, the increased thrombocytopenia observed with niraparib in mBRCA1/2 ovarian cancer cannot be sufficiently explained by primary pharmacology alone, but can potentially be attributed to the SERT and NET inhibition by niraparib alone since the SERT and NET reuptake inhibitor (sibutramine) has been shown to produce a prothrombotic state by altering the ultrastructure of platelets and fibrin networks.7,36 Similarly, the higher incidence of grade ≥3 anemia with talazoparib versus olaparib in breast cancer patients can potentially be attributed not only to the primary pharmacology, 19 which includes superior PARP trapping affecting the BM cell viability, but also to the comparatively long systemic exposure of talazoparib versus olaparib (elimination half-life of 90 hour versus 15 hour) that likely prolongs the regeneration of the erythroid compartment (normal, 3 weeks).4,6,15 In fact, the individual labels show that the higher incidence of talazoparib-induced anemia generally manifests around Days 21-24 following the initiation of treatment.4,6
The comparison of nonhematologic adverse events in ≥10% mBRCA1/2 ovarian and breast cancer patients demonstrates that the toxicities can likely be attributed to the differential off-target secondary pharmacology activities and broader PARP enzyme inhibition, at ≤ tCavg and/or tCmax exposure concentrations (Tables 2 and 3). Higher incidences of anxiety and hypertension with niraparib may be attributed to inhibition of triple-reuptake transporters, whereas depression with rucaparib is potentially due to inhibition of non-selective sigma receptors.37,38 Similarly, the higher incidences of insomnia, nasopharyngitis, dyspnea, and increased transaminase seen with niraparib versus olaparib are potentially due to the inhibition of triple reuptake transporters, whereas similar symptoms with rucaparib are potentially due to the inhibition of monoamine oxidase (MAO)-B. The higher incidences of rash with both rucaparib and niraparib versus olaparib may be due to inhibition of PIM3 kinase.39,40 Alopecia associated with talazoparib may be caused by a combination of inhibition of tankyrase 1 and/or 2 (PARP5A and 5B) activities in the hair follicle crypt cells and the long half-life of talazoparib.41,42 Despite the meaningful inhibition of both tankyrases at the respective clinical exposures (Figure 1), alopecia is not observed with olaparib and rucaparib. Therefore, alopecia is a potentially differentiating toxicity for talazoparib.
However, increased incidences of diarrhea are seen with all three inhibitors (not dose limiting) likely due to the direct inhibition of the tankyrases in intestinal crypt cells. In fact, diarrhea was a common treatment-related AE in the first-in-human study for E7449, a dual potent PARP1/2 and TNKS inhibitor, and inhibition of hair regrowth was observed in mice in a nonclinical study of E7449.41,43
Given that use of PARPi is being expanded beyond germline mBRCA1/2 cancers, our study comparing the safety effects of PARPi in healthy donor hBMMNCs (no germline DDR mutations) demonstrates that only olaparib and niraparib induced cytotoxicity at < tCavg concentrations (safety margin 0.4x), whereas the IC50 values for talazoparib and rucaparib were > tCavg with higher safety margins. The data also demonstrate that despite the potent PARP trapping activity, talazoparib does not induce substantial cytotoxicity to the dividing hBMMNC at clinically relevant concentrations and is therefore not a frank bone marrow toxicant as has been suggested previously. 19
Efficacy evaluation of the PARPi (–/+ TMZ) demonstrated that the H1048 SCLC cell line, which has mATR, mTP53, and mHDAC2, was most sensitive to the PARPi (cell viability IC50 ≤ tCavg) suggesting a potential benefit from a combination evaluation of ATR inhibitor with PARPi in SCLC. Synergistic efficacy of ATR inhibitor with PARPi has been demonstrated in prostate cancer cell lines with ATM loss and in ovarian cancer models.44-46 Alternately, in the DU145 cell line, which had several DDR mutations (mBRCA1 and 2, mFANCl, and mMLH1), only talazoparib was efficacious in decreasing cell growth (50% compared with vehicle) at ≤ tCavg concentration. Although the functional consequences of PARP trapping (S-phase accumulation, S-phase DSB, and early apoptosis) were not detected with talazoparib alone in DU145 cells, increasing PARP trapping with the low dose of TMZ was sufficient to left-shift all the responses to ≤ tCavg concentration. Despite the presence of multiple DDR mutations, only high-dose TMZ affected the functional PARP trapping endpoints and the cell growth decrease at ≤ tCavg for the other three PARPi, thereby demonstrating higher activity for talazoparib in androgen receptor inhibitor–resistant DU145 mCRPC cells. Finally, our data in the TK-6 lymphoblastoid cell line, which is expected to be sensitive to TMZ (MGMT-negative status), suggests potential combination opportunities for PARPi (+ TMZ) in certain hematologic malignancies and MGMT-negative cancers although the TMZ dose may require careful evaluation to determine the optimal therapeutic combination for the different PARPi.
Our study suggests that expanded use of PARPi in combination with PARP trappers and/or other DDR pathway inhibitors (eg, ATR inhibitor) could be a promising concept to evaluate in SCLC, advanced mCRPC, and hematologic malignancies. However, combinations should be carefully tested to achieve the optimum therapeutic index. Given the data in DU145 cells, use of talazoparib in combination with low dose TMZ may also be a promising therapeutic option to evaluate in advanced and hard to treat mCRPC. Our data also suggests that TMZ can potentially sensitize PARP inhibitors in MGMT-negative cancers in a combination setting.
List of Abbreviations
5-HT4, 5-hydroxytryptamine receptor 4 (non-selective sigma receptor; HTR4); ACC, activity concentration at cut-off; ADP, adenosine biphosphate; AE, adverse event. AIC, Akaike information criterion; ALT, alanine transaminase; APC, allophycocyanin; ARi, androgen receptor inhibitor; AST, aspartate transaminase; ATCC, American Type Culture Collection; ATM, ataxia–telangiectasia mutated; ATP, adenosine triphosphate; ATR, ataxia–telangiectasia and Rad3 related; AUC0–24, area under the concentration-time curve from time 0 to 24 hours; BID, twice daily; BRCA1/2, breast cancer susceptibility gene 1 and 2 (BRCA1/2 DNA repair associated); CC-3, cleaved caspase-3; CCM, Complete Culture Medium; CDK, cyclin-dependent kinase; DAT, dopamine transporter; DDR, DNA damage response; DLT, dose-limiting toxicity; DMEM, Dulbecco’s Modified Eagle’s Medium; DMSO, dimethyl sulfoxide; DSB, double-strand DNA breaks; DYRK1A/1B, dual specificity tyrosine phosphorylation regulated kinase 1A/1b; FACS, fluorescence-activated cell sorting; FANCl, FA complementation group L; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; Flt3, Fms-related receptor tyrosine kinase 3; γH2AX, phosphorylated H2A.X variant histone; gBRCAm, germline BRCA-mutated; hBMMNC, human bone marrow mononuclear cell; HDAC2, histone deacetylase 2; HER2, human epidermal growth factor receptor 2 (erb-b2 receptor tyrosine kinase 2); HRP, horseradish peroxidase; IC50, half maximal inhibitory concentration; Ki, inhibitory constant; mBC, metastatic breast cancer; mBRCA1/2, BRCA1/2 mutation-positive; MAO, monoamine oxidase; mCRPC, metastatic castration-resistant prostate cancer; MGMT, O-6-methylguanine-DNA methyltransferase; MLH1, mutL homolog 1; MMS, methyl methane sulphonate; NA, not active; ND, not determined; NET, norepinephrine transporter; PARP, poly(ADP-ribose) polymerase; PARPi, poly(ADP-ribose) polymerase inhibitor(s); PASK, PAS domain containing serine/threonine kinase; PBS, phosphate buffered saline; PE, phycoerythrin; PI, propidium iodide; PIM3, Pim-3 proto-oncogene, serine/threonine kinase; PMT, photo multiplier tube; PoD, point-of-departure; PPB, parts per billion; QD, once daily; RRID, Research Resource Identifier; SCLC, small cell lung cancer; SERT, serotonin transporter; tCavg, total concentration average; tCmax, total concentration maximum; tcpl, ToxCast pipeline; TMZ, temozolomide; TNKS, tankyrase; TP53, tumor protein p53
Supplemental Material
Supplemental Material - Comparative Analyses of Poly(ADP-Ribose) Polymerase Inhibitors
Supplemental Material for Comparative Analyses of Poly(ADP-Ribose) Polymerase Inhibitors by Mausumee Guha, Zhanna Sobol, Mathew Martin, Michelle Hemkens, Tae Sung, Elizabeth Rubitski, Richard Spellman, Martin Finkelstein, Nasir Khan, and Wenyue Hu in International Journal of Toxicology
Footnotes
Acknowledgments
Editorial support was provided by Bethany Delcuze, PhD, Hannah Logan, PhD, and Daniela DiBiase, MS MPH, of CMC AFFINITY, a division of IPG Health Medical Communications and was funded by Pfizer.
Declaration of Conflict(s) of Interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are current or former employees of Pfizer Inc. and may own stock or stock options in Pfizer.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Pfizer Inc.
Consent for Publication
All authors have consented for the manuscript to be published.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.