
Abstract
Opioid-induced constipation (OIC) and other gastrointestinal (GI) symptoms of opioid-induced bowel dysfunction (OIBD) significantly deteriorate patients’ quality of life and may lead to noncompliance with opioid schedule and undertreatment of pain. Although traditional oral laxatives are the first-line treatment of OIC, they do not address OIBD pathophysiology, and display numerous adverse effects. OIC treatment includes prokinetics (lubiprostone), opioid switch, and changing route of opioid administration. Targeted management of OIBD comprises the use of purely peripherally acting μ-opioid receptor antagonists (PAMORA): naloxegol and methylnaltrexone. Naloxegol (NKTR-118) is a polymer conjugate of the opioid antagonist naloxone. The polyethylene glycol limits naloxegol capacity to cross the blood–brain barrier (BBB). Naloxegol is substrate for the P-glycoprotein (P-gp) transporter. The central nervous system penetration of naloxegol is negligible due to reduced permeability and its increased efflux across the BBB, related to P-gp transporter. Naloxegol antagonizes μ- and κ-opioid receptors and displays low affinity to δ-opioid receptors in the GI tract, thereby decreasing OIBD symptoms without reversing central analgesic effects. Naloxegol is metabolised through CYP3A4 to six metabolites, with the majority of the dose (68%) excreted with faeces and less (16%) with urine. The dose of naloxegol equals 25 mg administered orally once daily on a fasting condition. Mild or moderate hepatic impairment has no impact on naloxegol dosing; naloxegol was not studied and is not recommended in patients with hepatic failure. Dose reduction (12.5 mg once daily) and caution is recommended in patients with moderate-to-severe renal impairment. Efficacy (bowel movement in 42–49% of patients not responsive to laxatives) and safety of naloxegol were confirmed in studies conducted in patients with OIC and nonmalignant pain. Naloxegol may be useful for cancer patients with OIC, although studies in this population are lacking.
Keywords
Introduction: opioid-induced bowel dysfunction
Opioids are often successfully used for pain management but they may also induce numerous and potentially serious adverse events (AEs). Although tolerance develops only for some of opioid AEs, such as sedation, there may be little or no tolerance development to opioid-induced gastrointestinal (GI) AEs. Therefore, patients should be closely monitored by the staff to avoid or decrease the intensity of opioid-induced AEs that may significantly affect patients’ quality of life (QOL) and lead to noncompliance with opioid regimens resulting in undertreatment of chronic pain [Cherny et al. 2001]. Some common opioid AEs are symptoms associated with the GI tract, collectively termed opioid-induced bowel dysfunction (OIBD).
OIBD symptoms reflect a complex impact of opioids on the GI tract. The most typical, and usually most burdensome symptom of OIBD is opioid-induced constipation (OIC). However, the impact of opioids on the whole GI tract also comprises such symptoms as dry mouth, gastro-esophageal reflux-related symptoms (heartburn), nausea, vomiting, chronic abdominal pain, bloating, constipation-related symptoms: straining, hard stools, painful, infrequent and incomplete bowel movements (BMs), and diarrhoea-related symptoms: urgency, loose and frequent BMs [Brock et al. 2012]. OIBD is a common complication of long-term opioid therapy and may lead to QOL deterioration and undertreatment of pain; it affects 40–80% of patients treated with opioids [Bell et al. 2009; Cook et al. 2008].
OIBD induces activation of peripheral µ-opioid receptors located in the gut wall with less central effects [Reimer et al. 2009]. Opioid receptors (predominantly µ and δ) are located in the submucosal plexus and in the myenteric plexus, respectively, and in immune cells in the lamina propria of the gut wall. The activation of µ-opioid receptors inhibits excitatory and inhibitory neural pathways within the enteric nervous system that coordinates motility. Inhibition of excitatory neural pathways decreases peristaltic contractions. In turn, the blockade of inhibitory neural pathways increases GI muscle activity and elevates resting muscle tone, spasm and nonpropulsive motility patterns. These mechanisms induce delayed gastric emptying and slow down the intestinal transit [Davis 2005; Holzer, 2007; Mori et al. 2013].
The effective management of patients with OIBD should encompass a meticulous clinical evaluation, including all OIBD symptoms, taking into account patients’ complaints and QOL. For this purpose, specific validated instruments for OIBD assessment may be useful [Olesen and Drewes, 2011]. Thus, a complex assessment and management that addresses the underlying causes and pathomechanism of OIBD is recommended.
Newer strategies of OIBD management comprise prokinetics (lubiprostone), purely peripherally acting μ-opioid receptor antagonists (PAMORAs) and a combination of opioid agonist and antagonist (prolonged-release oxycodone/naloxone). The latter may also be considered as a preventive measure of OIBD development in patients who require chronic opioid administration. Treatment with PAMORA may be used in patients with OIBD, especially when traditional laxatives fail.
Naloxegol
Pharmacodynamics
Naloxegol (NKTR-118) is a polymer conjugate of the opioid antagonist naloxone. The chemical structure of naloxegol is shown on Figure 1. The polyethylene glycol moiety (PEG) limits naloxegol capacity to cross the blood–brain barrier. Naloxegol thus belongs to PAMORA. Naloxegol is indicated for the treatment of adult patients with OIC who have had an inadequate response to laxative(s) [AstraZeneca UK Ltd, 2016]. When administered at the recommended dosing, naloxegol antagonizes the μ-receptor in the GI tract, thereby decreasing the constipating effects of opioids. In-vitro studies have demonstrated significant affinity for the μ- or κ-opioid receptors (inhibition constant, Ki = 7.42nmol/l and Ki = 8.65 nmol/l, respectively) and low affinity (Ki = 203.0 nmol/l) for the δ-opioid receptors [AstraZeneca UK Ltd, 2014]. The presence of the PEG moiety in naloxegol reduces its passive permeability as compared with naloxone. PEGylation makes naloxegol a substrate for the P-glycoprotein (P-gp) transporter. Due to the reduced permeability and increased efflux of naloxegol across the blood–brain barrier, related to P-gp transporter, the central nervous system (CNS) penetration of naloxegol is negligible and it reduces OIC in the GI tract without reversing the central analgesic effect.
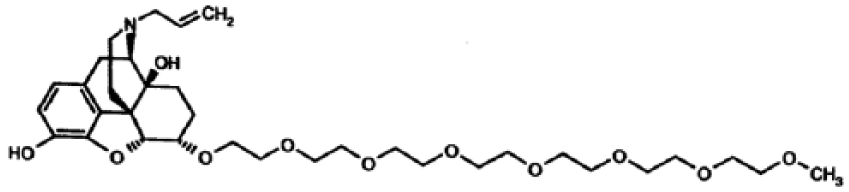
Chemical structure of naloxegol.
Naloxegol works as an antagonist primarily at the μ-opioid receptor. In preclinical models, permeability at the blood–brain barrier was reduced 15-fold compared with naloxone. Moreover, naloxegol was found to attenuate the gastric peristaltic effects of morphine without a significant effect on analgesia [Eldon et al. 2007; Neumann et al. 2007a]. Morphine-induced miosis was used to evaluate the ability of naloxegol to cross the blood–brain barrier. Healthy male volunteers were given morphine 5 mg/70 kg intravenously along with oral naloxegol (8–1000 mg) or placebo solution using a double-blind, crossover design with a 1-week washout period. No central antagonism of pupil constriction was observed in 46 (95.8%) participants. Two participants had a possible attenuation; one received naloxegol 250 mg, and the other received naloxegol 1000 mg along with the morphine dose. Naloxegol shortened oral–caecal transit time, prolonged by morphine more than placebo (p < 0.05), with a 50% effective dose that was ≈ 15 mg [Neumann et al. 2007b]. Although the study was conducted with naloxegol solution, it was demonstrated to be bioequivalent to naloxegol tablets [Odinecs et al. 2009].
Naloxegol does not significantly affect the QT interval at therapeutic (25 mg) and supratherapeutic (150 mg) doses in healthy volunteers, therefore it has no relevant effect on cardiac repolarization in patients with OIC. Moreover, naloxegol has no impact on heart rate, blood pressure and QRS intervals and T-wave morphology [Gottfridsson et al. 2013].
Pharmacokinetics
Naloxegol undergoes rapid absorption after oral administration, with peak plasma concentration (Cmax) achieved in less than 2 hours. Due to enterohepatic recycling, the second peak occurs in most patients 0.4–3 hours after the first peak. Since naloxegol is a PEGylated analogue, it stays longer in circulation and is metabolized predominantly in the liver by the CYP3A4 enzyme; it is also a substrate of the P-gp transporter. Its plasma-protein binding equals 4.2%. Naloxegol plasma half-life (T1/2) extends from 6 to 11 hours and it is primarily excreted with faeces. The metabolites (six in number) are predominantly excreted in faeces (67%) and the rest in the urine. These pharmacokinetic parameters have been confirmed in a study conducted in healthy subjects [Bui et al. 2015].
The pharmacokinetic profile of naloxegol is well characterized in healthy subjects, in patients with hepatic or renal impairment, and in patients with OIC. Naloxegol exhibits dose-proportional pharmacokinetics in healthy subjects and patients with OIC and is rapidly absorbed following oral administration, with a similar half-life across all dose levels. The role of the hepatobiliary system in the metabolism of naloxegol is well established, with biliary excretion as the primary route of elimination; however, surprisingly, mild or moderate hepatic impairment had minimal effect on the pharmacokinetics of naloxegol, whereas increased exposures were observed in a small number of patients with moderate or severe renal impairment. The absolute bioavailability of naloxegol in humans has not been determined yet. Factors such as age, sex, ethnicity, body weight, prior response to laxatives, and baseline opioid-analgesic dosing have minimal impact on the pharmacokinetics of naloxegol. However, administration of naloxegol with food is associated with increased bioavailability. In a study of human volunteers, there was a 45% increase in area under the plasma concentration curve (AUC) and a 30% increase in Cmax associated with the administration of naloxegol with a high-fat meal, therefore it is recommended that naloxegol is administered in the fasting state [Bui et al. 2015].
Naloxegol undergoes hepatic metabolism via cytochrome P-450 3A4 enzyme and elimination by biliary excretion. The renal system plays a minimal role in metabolism. In human volunteers, radioactivity occurred primarily in the faeces (67.7%) and urine (16%). Naloxegol undergoes enterohepatic recycling, as suggested by the bimodality of AUC [Van Paaschen et al. 2008]. A recent controlled study conducted in healthy volunteers demonstrated that an addition of quinidine (a strong P-gp inhibitor and a weak CYP3A4 inhibitor) to naloxegol and morphine did not increase naloxegol distribution to the CNS and has not influenced morphine pharmacokinetics. An increase of naloxegol AUC and Cmax values was attributed to an inhibitory effect of quinidine on naloxegol metabolism [Bui et al. 2016].
A population pharmacokinetics of naloxegol was evaluated in 1247 healthy volunteers and patients recruited from 14 phase I, IIb and III studies. In order to assess the clinical relevance of covariate effects on the pharmacokinetic profile of naloxegol, simulations using the 25 mg daily dosing regimen were conducted. Concomitant administration of naloxegol with strong CYP3A4 inducers decreased systemic exposure by 90%, while CYP3A4 strong inhibitors resulted in an 8-fold increase in naloxegol systemic exposure. Moderate CYP3A4 inhibitors increased AUC and Cmax by 60% and 30%, respectively. Concomitant administration of naloxegol with P-gp inducers resulted in reduced naloxegol systemic exposure by 60–70%, and P-gp inhibitors increased systemic exposure by 40–50%. Black patients showed a 20% decrease in AUC and a 10% decrease in Cmax, and Asian patients had a 30% increase in Cmax, compared with other races, most probably associated with CYP3A5 polymorphism. Administration of strong CYP3A4 inhibitors or inducers had a clinically relevant influence on naloxegol exposure; the effects of moderate CYP3A4 inhibitors display significant intersubject variability [Al-Huniti et al. 2016].
Mild–moderate hepatic impairment has been found to have no significant effect on naloxegol metabolism and clearance, and there is no need for dose reduction. Naloxegol was not studied in patients with hepatic failure, thus there is no recommendation in this patient population [Bui et al. 2014a]. Mild renal impairment has no impact on naloxegol pharmacokinetics. However, patients with creatinine clearance less than 60 ml/min are started off with a lower dose of 12.5 mg once daily and if well tolerated, can be switched over to a normal dose (25 mg once daily) [Bui et al. 2014b]. Naloxegol has been assigned category C status for use in pregnancy. Carcinogenicity studies performed to date demonstrated an increase in Leydig cell adenomas and interstitial cell hyperplasia, but at a dose in excess of a human dose. The studies in suckling rats demonstrated the secretion of naloxegol in milk [AstraZeneca UK Ltd, 2016].
Drug interactions
The concentration and activity of naloxegol are increased by the concurrent use of CYP3A4 inhibitors and reduced by CYP3A4 inducers. Concurrent use with a strong CYP3A4 inhibitor (e.g. clarithromycin, itraconazole) is contraindicated, and the use with a moderate CYP3A4 inhibitor (e.g. diltiazem, verapamil) should be avoided whenever possible or used with caution. It has been shown that the significant interaction of naloxegol with drugs altering CYP3A4/P-gp transporter is possible, hence the use of naloxegol concurrently with drugs that are strong CYP3A4 inhibitors should be avoided. When moderate CYP3A4 inhibitors are concurrently administered, the naloxegol dose should be reduced to 12.5 mg, administered once daily. Any dose adjustment is not recommended for a concurrent use of weak CY3A4 inhibitors. The consumption of grapefruit products should also be avoided during a treatment with naloxegol. The concurrent use of a strong CYP3A4 inducer (e.g. carbamazepine, rifampicin, St John’s wort) is not recommended. The use of naloxegol with another opioid antagonist should be avoided because of the increased risk of opioid withdrawal. Maintenance laxative therapy should be discontinued before initiating treatment with naloxegol. However, if there is a suboptimal response to naloxegol after 3 days, laxatives can be used as needed [AstraZeneca, 2014].
Dosing guidelines
Naloxegol is administered at a dose of 25 mg once daily on an empty stomach at least 30 minutes (in Europe) or at least 1 hour (in the US) before the first meal of the day or 2 hours after the first meal of the day. The tablets should be swallowed whole; they should not be chewed or crushed. Avoiding consumption of grapefruit or grapefruit juice with the naloxegol dose is recommended. When naloxegol started, it is recommended that all currently used maintenance laxative therapy should be ceased, until clinical effect of naloxegol is determined.
The starting dose is 12.5 mg once daily for individuals with creatinine clearance less than 60 ml/min and increased up to 25 mg once daily if well tolerated. The dosage should be reduced to 12.5 mg once a day in patients with moderate, severe, or end-stage renal impairment and in patients in whom the concurrent use of a moderate CYP3A4 inhibitor is unavoidable. If the treatment with the opioid analgesic is discontinued, naloxegol should also be discontinued.
Naloxegol is contraindicated in patients with hypersensitivity to the drug, any excipients and other PAMORAs, and those using strong CYP3A4 inhibitors. Cancer patients with symptoms of bowel obstruction and those with increased risk of GI perforation [GI or peritoneal tumours, recurrent or advanced ovarian cancer and patients treated with vascular endothelial growth factor (VEGF) inhibitors] should also be excluded from naloxegol treatment. Caution is recommended in patients concurrently treated with methadone, those with impaired blood–brain barrier (primary brain tumours or metastases to brain, vascular diseases of CNS), increased risk of GI perforation, cardiovascular diseases and those treated with CYP3A4 inducers. Symptoms of opioid withdrawal, severe intensity of abdominal pain and diarrhoea suggest discontinuation of naloxegol treatment. The efficacy and safety of naloxegol in the paediatric population has not been established so far, thus, the drug is not recommended in children.
Due to the differences between the agents of the PAMORA, it is worth comparing the available drugs in this class. At present, there are three medications on the pharmaceutical market that can be used for similar indications but may vary from one another in terms of their safety profile, which should be taken into account when choosing a drug [Nelson and Camilleri, 2016]. Naloxegol is the first oral PAMORA for OIC in adults with chronic noncancer pain, with approval from the US Food and Drug Administration and the European Union; in the latter, also, for patients with cancer-related pain, although the study in this population failed to recruit enough patients [Von Roenn et al. 2013].
Overview of naloxegol clinical studies
In a phase II, randomized, double-blind, placebo-controlled, dose-escalation study, the efficacy and safety of three dose levels of oral naloxegol in the treatment of OIC in patients with nonmalignant or cancer-related pain was evaluated [Webster et al. 2013]. Eligible patients with OIC (n = 207), defined as less than three spontaneous bowel movements (SBMs) per week with accompanying symptoms, on a stable opioid regimen of 30–1000 mg/day morphine equivalents for at least 2 weeks, were randomized to receive 4 weeks of double-blind placebo or naloxegol (5 mg, 25 mg, or 50 mg) once daily in sequential cohorts after a 1-week placebo run-in.
The primary endpoint, median change from baseline in SBMs per week after week 1 of drug administration, was statistically significant for the 25 mg and 50 mg naloxegol cohorts versus placebo: 2.9 versus 1.0 (p = 0.002) and 3.3 versus 0.5 (p = 0.0001), respectively. The increase in SBMs versus placebo was maintained over 4 weeks for naloxegol 25 mg: 3.0 versus 0.8 (p = 0.0022) and 50 mg: 3.5 versus 1.0 (p < 0.0001). Naloxegol was generally well tolerated across all dosages. The most frequent AEs were abdominal pain, diarrhoea, and nausea. Most AEs at 5 mg and 25 mg/day were mild and transient. Similar AEs occurred with increased frequency and severity in the 50 mg cohort. There was no evidence of a statistically significant increase from baseline in pain, opioid use for the 25 and 50 mg cohorts, or centrally mediated opioid withdrawal signs with or without symptoms from naloxegol. Naloxegol improved the frequency of SBMs compared with placebo and was generally well tolerated in this population of patients with OIC. The dose of 25 mg naloxegol was chosen for further exploration in phase III studies [Webster et al. 2013].
Two phase III, double-blind clinical studies (KODIAC-04 and KODIAC-05) have been completed and demonstrated significant increase in SBMs compared with placebo for the 25 mg naloxegol dose (in one study, also for the 12.5 mg dose) [Chey et al. 2014]. In both studies (study 04, 652 participants; study 05, 700 participants), outpatients with noncancer pain and OIC were randomly assigned to receive a daily dose of 12.5 mg or 25 mg of naloxegol or placebo. The primary endpoint was the 12-week response rate (⩾3 SBMs per week and an increase from baseline of ⩾1 SBM for ⩾9 of 12 weeks and for ⩾3 of the final 4 weeks) in the intention-to-treat population. The key secondary endpoints were the response rate in the subpopulation of patients with an inadequate response to laxatives before enrollment, the time to first postdose SBM, and the mean number of days per week with one or more SBMs.
Response rates were significantly higher with 25 mg of naloxegol than with placebo (intention-to-treat population: study 04, 44.4% versus 29.4%,p = 0.001; study 05, 39.7% versus 29.3%, p = 0.02; patients with an inadequate response to laxatives: study 04, 48.7% versus 28.8%, p = 0.002; study 05, 46.8% versus 31.4%, p = 0.01); in study 04, response rates were also higher in the group treated with 12.5 mg of naloxegol (intention-to-treat population, 40.8% versus 29.4%, p = 0.02; patients with an inadequate response to laxatives, 42.6% versus 28.8%, p = 0.03) (Table 1). A shorter time to the first postdose SBM and a higher mean number of days per week with one or more SBMs were observed with 25 mg of naloxegol versus placebo in both studies (p < 0.001), and with 12.5 mg of naloxegol in study 04(p < 0.001). Pain scores and daily opioid dose were similar among the three groups. AEs (primarily gastrointestinal) occurred most frequently in the groups treated with 25 mg of naloxegol. Treatment with naloxegol, as compared with placebo, resulted in a significantly higher rate of treatment response, without reducing opioid-mediated analgesia [Chey et al. 2014].
Efficacy of naloxegol (response rates, relative risks, p values and number needed to treat) in phase III study Chey et al. [2014].
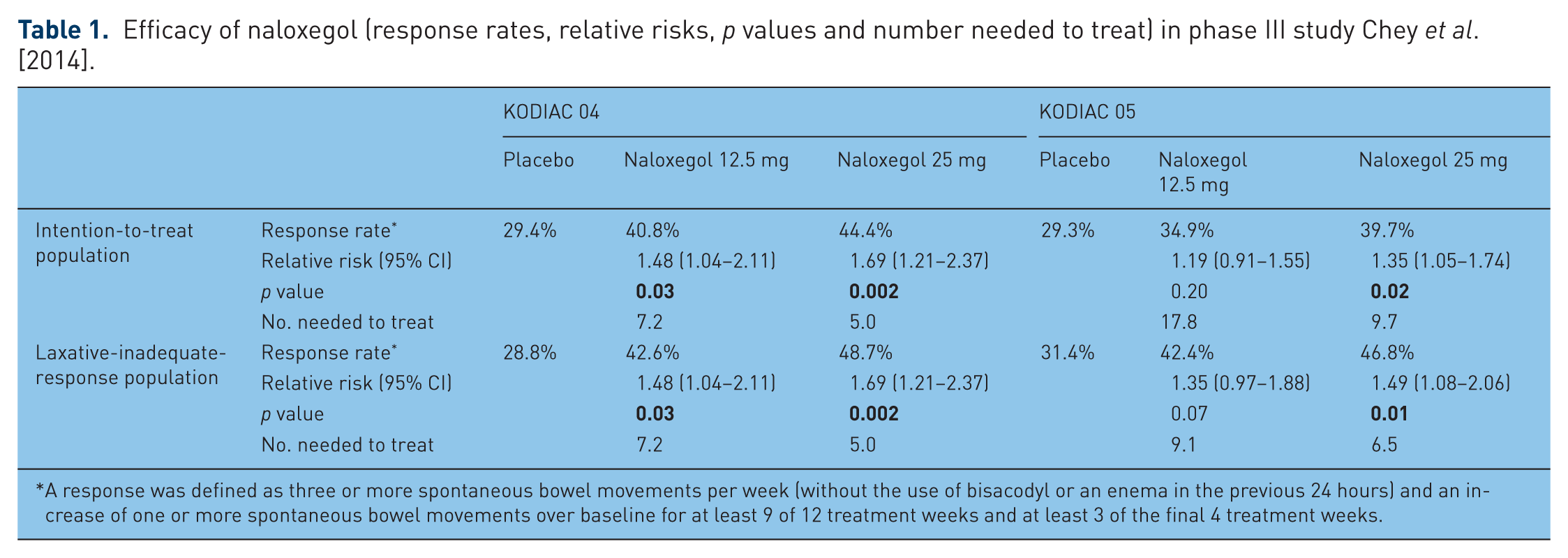
A response was defined as three or more spontaneous bowel movements per week (without the use of bisacodyl or an enema in the previous 24 hours) and an increase of one or more spontaneous bowel movements over baseline for at least 9 of 12 treatment weeks and at least 3 of the final 4 treatment weeks.
A 52-week, multicentre, open-label, randomized, parallel-group phase III study was conducted in outpatients with nonmalignant pain taking 30–1000 morphine-equivalent units per day for at least 4 weeks. Patients were randomized 2:1 to receive naloxegol 25 mg/day or usual care [(UC) investigator-chosen laxative regimen] treatment for OIC [Webster et al. 2014]. The safety set comprised 804 patients (naloxegol, n = 534; UC, n = 270).
Mean exposure duration was 268 days with naloxegol and 297 days with UC. Pain scores and mean daily opioid doses remained stable throughout the study; no attributable opioid withdrawal AEs was observed. Two patients in each group had an adjudicated major adverse cardiovascular event unrelated to study drug; no AEs were reported nor adjudicated as bowel perforations. Naloxegol at a dose of 25 mg daily administered up to 52 weeks was safe and quite well tolerated in patients with nonmalignant pain and OIC [Webster et al. 2014].
A more recent analysis of KODIAC-04 and KODIAC-05 studies demonstrated benefits of naloxegol in patients with inadequate response to conventional laxatives (LIR) [Tack et al. 2015]. Patients with LIR were defined as those who, during 2 weeks, had to have reported OIC symptoms of at least moderate severity while taking at least one laxative class for a minimum of 4 days. Outpatients with noncancer pain and OIC were randomly assigned to receive a daily dose of 12.5 mg (n = 240) or 25 mg (n = 241) of naloxegol or placebo (n = 239).
In general, positive findings observed in both studies were confirmed in the analysis of LIR patients. OIC response rates for the naloxegol 25 mg (p < 0.001) and the 12.5 mg (p < 0.005) LIR dose groups were significantly higher than in the case of placebo. Median times to first postdose SBM were 7.6, 19.2 and 41.1 hours for the naloxegol 25 mg, naloxegol 12.5 mg, and placebo groups, respectively. Other SBM measures, daily symptoms of OIC, and both the Patient Assessment of Constipation Symptoms, and Patient Assessment of Constipation Quality of Life scores improved from baseline with naloxegol treatment. Changes from baseline in opioid dose, pain scores and opioid withdrawal scores were similar among the treatment groups. These results suggest that naloxegol is an effective and well tolerated option in the treatment of OIC in patients who do not achieve adequate response from traditional laxatives [Tack et al. 2015].
A summary of naloxegol studies is presented in Table 2 [Bruner et al. 2015].
Overview of naloxegol studies, adapted from Bruner et al. [2015], modified.

AE, adverse event; AUC, area under the plasma concentration curve; SBMs, spontaneous bowel movements; Cmax, maximum plasma concentration; DB, double blind; eGFR, estimate glomerular filtration rate; ESRD, end-stage renal disease; GI, gastrointestinal; IHD, intermittent hemodialysis; MDRD, modification of diet in renal disease; OIC, opioid-induced constipation; PO, by mouth; H, history; incl., including; M, months; RI, renal impairment; OME, oral morphine equivalent; pts, patients; PE, Primary endpoint; SE, Secondary endpoint; LIR, inadequate response to laxatives; y, years old.
Adverse events
Naloxegol is relatively well tolerated, although it may induce AEs, mostly from the GI tract, such as abdominal pain, diarrhoea, nausea, flatulence, upper abdominal pain and vomiting (Table 3) [Chey et al. 2014]. These effects are usually transient and their intensity mostly mild to moderate. Other naloxegol AEs comprise fall, headache and hyperhydrosis. Naloxegol does not induce opioid withdrawal nor impair analgesia during opioid treatment. There were no reports on GI perforation, although patients with risk groups were not enrolled into the phase III studies [Chey et al. 2014].
The most common gastrointestinal adverse events (in percentages) in phase III study of naloxegol conducted among patients with noncancer pain Chey et al. [2014].

In a phase II study [Webster et al. 2013], the most frequently reported treatment-emergent AEs were GI complaints and included abdominal pain, diarrhoea and nausea. Similarly, the most frequently occurring treatment-emergent AEs considered by the investigator to be related to study medication were GI symptoms and these AEs increased with increments of naloxegol doses from 13 (39.4%) in the 5 mg cohort to 14 (46.7%) in the 25 mg cohort, and to 21 (60.0%) in the 50 mg cohort. The most common GI AEs in patients receiving naloxegol 5 mg, 25 mg and 50 mg doses were as follows: diarrhoea, five (15.2%), four (13.3%) and 11 (31.4%) patients; nausea, five (15.2%), four (13.3%) and seven (20.0%) patients; upper abdominal pain, six (18.2%), three (10.0%), and 10 (28.6%) patients; and abdominal pain, one (3.0%), nine (30.0%) and six (17.1%) patients, respectively [Webster et al. 2013].
Hyperhidrosis occurred in four (12.1%) patients receiving 5 mg of naloxegol, two (6.7%) patients receiving 25 mg naloxegol, and three (8.6%) patients receiving 50 mg of naloxegol. Treatment-emergent AEs led to study discontinuation of three patients in the 5 mg cohort (placebo, two; naloxegol, one), three patients in the 25 mg cohort (placebo, zero; naloxegol, three), and 14 patients in the 50 mg cohort (placebo, three; naloxegol, eleven). In the 50 mg cohort, seven cases of premature study discontinuation due to treatment-emergent AEs were considered possibly or probably related to study medication and included abdominal pain, diarrhoea, nausea, vomiting, and abdominal cramping [Webster et al. 2013].
In a phase III study AEs leading to discontinuation in at least three patients in the 25 mg group were diarrhoea (2.8% of patients), abdominal pain (1.9%), and upper abdominal pain (1.4%) in study 04 and abdominal pain (3.9%), diarrhoea (3.4%), nausea (1.7%), and vomiting (1.7%) in study 05. Individual serious AEs were infrequent and similar in type and frequency across the three groups in both studies. Serious AEs appeared in 5.2% and 6.1% (naloxegol 12.5 mg), in 3.3% and 3.4 % (naloxegol 25 mg) patients in studies 04 and 05, respectively. No individual event was reported in more than two patients in any group in either study. Cardiovascular events were infrequent: acute myocardial infarction was observed in one patient in both naloxegol groups in 04 study and in two patients from placebo group in 05 study; in one patient from the latter group this AE was considered to be related to the study drug [Chey et al. 2014].
In the long-term study during 52 weeks of the treatment, naloxegol was generally well tolerated [Webster et al. 2014]. The frequency of AEs was 81.8% with naloxegol and 72.2% with UC. AEs occurred more frequently for naloxegol versus UC, such as abdominal pain (17.8% versus 3.3%), diarrhoea (12.9% versus 5.9%), nausea (9.4% versus 4.1%), headache (9.0% versus 4.8%), flatulence (6.9% versus 1.1%) and upper abdominal pain (5.1% versus 1.1%). Most naloxegol-induced GI AEs occurred early, resolving during or after naloxegol discontinuation and were mild or moderate in severity. AEs leading to discontinuation of naloxegol appeared in 56 (10.5%) patients; the most common were as follows: diarrhoea, 11 (2.1%), abdominal pain, nine (1.7%) and vomiting in five (0.9%) patients. In the group UC, five (1.8%) patients withdrew from the study due to AEs [Webster et al. 2014].
The pooled analysis of LIR patients [Tack et al. 2015] confirmed findings from 04 and 05 controlled studies [Chey et al. 2014]. A greater incidence of AEs was found in naloxegol 25 mg group (63.1%) compared with 12.5 mg group (50.6%) and placebo (50.0%). The most frequent AEs reported by LIR patients receiving naloxegol 12.5 mg and 25 mg, respectively, were abdominal pain (7.6% and 15.8%), diarrhoea (6.8% and 10.4%), nausea (6.3% and 8.3%), vomiting (2.5% and 3.7%) and fall (2.5% and 0.8%). Frequency of flatulence, upper abdominal pain and hyperhidrosis were higher in naloxegol 25 mg compared with 12.5 mg dose and placebo [Tack et al. 2015].
Summary
OIBD is a common complication in patients receiving long-term treatment with opioids. Any intervention should always be preceded with a detailed assessment of the OIBD symptoms in the wider context of the underlying disease and comorbidities as well as thorough patient’s assessment, including nonmedical problems [Poulsen et al. 2014]. Therapeutic choices can be made based on results of the treatment evaluated by the Bowel Function Index (BFI) as scores less than 30 indicate normal bowel function and BFI results 30 or higher suggest constipation [Argoff et al. 2015]. More clear definition of OIC may allow for more precise comparisons between different interventions [Gaertner et al. 2015].
A significant progress has been made with targeted therapies for the management of patients with the symptoms of OIBD [Brenner and Chey, 2014; Camilleri et al. 2014; Ford et al. 2013]. Among currently available PAMORAs, naloxegol seems to be the most convenient thanks to an oral route and once-daily administration [Poulsen et al. 2015]. Naloxegol is effective in patients with OIC not responding to traditional laxatives [Garnock-Jones, 2015]. It seems to display a well-tolerated profile, as severe AEs were not reported regarding bowel perforation that was noticed in seven patients treated with subcutaneously administered methylnaltrexone [Mackey et al. 2010]. However, clinical studies of methylnaltrexone did not report such AEs and confirm its efficacy in patients with OIC [Siemens and Becker, 2016].
Apart from orally administered traditional oral laxatives and PAMORAs, currently available therapeutic options for patients with OIC comprise oxycodone/naloxone (restricted by a maximal daily dose of 160 mg/80 mg, normal hepatic function and intact portal circulation, although in the US, oxycodone/naloxone is approved only as an abuse-deterrent formulation) [Ahmedzai et al. 2015] and lubiprostone (the only prokinetic registered for the treatment of OIC) [Jamal et al. 2015; Cryer et al. 2014]. However, lubiprostone does not address the underlying OIBD pathophysiology and the drug is not recommended for patients treated with diphenyl heptane opioids, such as methadone, as they interfere with the activation of chloride channels [Cuppoletti et al. 2013]. Opioid switch or change of route of opioid administration may also be considered although the evidence is limited. The final steps are rectal measures and manual stool evacuation (under analgesia and sedation) when all measures fail [Leppert, 2015].
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.