
Abstract

Introduction
Numerous studies have addressed the relationship between cancer and venous thromboembolism (VTE) dating back to 1865 [Trousseau, 1865]. An increased prevalence of occult cancer has been noted in patients presenting with idiopathic VTE. The same applies for occult cancer in patients with secondary and idiopathic VTE [Prandoni et al. 1992].
The use of anticancer therapy (chemotherapy/hormone) can increase the risk of thromboembolic diseases in cancer patients. Chemotherapeutic agents can activate the clotting cascade through the release of procoagulant fibrinolytic substances by tumor cells, or through endothelial damage or the stimulation of tissue factor production by host cells [Shaib et al. 2010]. This hypercoaguable state is further aggravated by the use of venous access catheters and possibly growth factors [Chu et al. 2014].
The thrombogenic effect of anticancer drugs has been best studied in breast cancer where cytotoxic chemotherapy and tamoxifen both appear independently to increase the risk of venous and arterial thrombosis.
Thrombogenicity (and/or hypercoagulopathy) has rarely been reported with 5-FU. There have been changes in protein C, fibrinopeptide A, von Willebrand factor, and fibrin D-dimer reported following 5-FU therapy in vivo and in vitro [Kuzel et al. 1990; Kinhult et al. 2001]. We recently published a case report of a DPYD variant (Y186C) specific to individuals of African descent in a patient with life-threatening 5-FU toxicity [Kinhult et al. 2003]. In the same patient, we observed factor VII (FVII) deficiency; a finding that we believe deserves further attention and discussion. 5-FU causes endothelial injury leading to a procoagulant state, which may be of importance in the underlying mechanism of 5-FU-induced cardiotoxicity.
Case report
The patient was a 60-year-old African-American woman with histological diagnosis of stage IV adenocarcinoma of the sigmoid colon with multiple metastatic sites in the liver, left supraclavicular and mediastinal lymphadenopathy, diffuse retroperitoneal and portacaval lymphadenopathy. The patient’s past medical history included hypertension, asthma, fibromyalgia, and bipolar disorder. The patient’s performance status was zero except uncontrolled hypertension (159/95 mmHg) for which she was increased on lisinopril from 10 to 20 mg daily. The laboratory data including complete blood count [white blood cell count of 8.7 K/µl (range 4.0–11.0 K/µl), absolute neutrophil count of 5.5 K/µl (range 1.5–7.5 K/µl), hemoglobin 12.1 g/dl (range 13.5-16.0 g/dl), and platelet count of 178 K/µl (range 150-400 K/µl)], chemistries [creatine of 0.9 mg/dl, blood urea nitrogen (BUN) of 20 mg/dl (range 6–24 mg/dl), albumin of 4.0 mg/dl (range 3.4–4.8 mg/dl)], liver function tests, and coagulation profile [prothrombin time (PT) of 11.1 s (range 9.7–13.6 s) and an international normalized ratio (INR) of 0.7 (range 0.9–1.1)] were within normal limits. Therefore, she was started on systemic therapy with FOLFOX (fluorouracil 400 mg/m2 IV push, fluorouracil 2400 mg/m2 IV over 46 hours, leucovorin 400 mg/m2, and oxaliplatin 85 mg/m2) with the plan to add bevacizumab once the poorly controlled hypertension was controlled.
Approximately 10 days following the first cycle of FOLFOX, the patient presented with pancytopenia [white blood cell count of 2.4 K/µl (range 4.0–11.0 K/µl), absolute neutrophil count of 1.2 K/µl (range 1.5–7.5 K/µl), hemoglobin 10.8 g/dl (range 13.5–16.0 g/dl), and platelet count of 80 K/µl (range 150–400 K/µl)], severe mucositis, esophagitis, inadequate pain control, and dehydration. The rest of the laboratory data showed: BUN of 34 mg/dl (range 6–24 mg/dl), potassium of 3.1 meq/l (range 3.6–5.1 meq/l), albumin of 3.3 mg/dl (range 3.4–4.8 mg/dl), phosphorus of 2.5 mg/dl (2.7–4.5 mg/dl), alkaline phosphatase of 146 IU/l (range 40–130 IU/l), and a lactate dehydrogenase (LDH) of 288 IU/l (range 50–175 IU/l). Coagulation panel revealed a PT of 14.1 s (range 9.7–13.6 s) and an INR of 1.3 (range 0.9–1.1). INR peaked at 3.1 (Figures 1–3). Mixing study was consistent with isolated functional FVII deficiency. Factors II, V, X were within normal limits. Repeat mixing study 3 days later showed the same distribution of factor functionally. INR improved to 1.6 with vitamin K administration but never normalized (Table 1 and Figure 2).
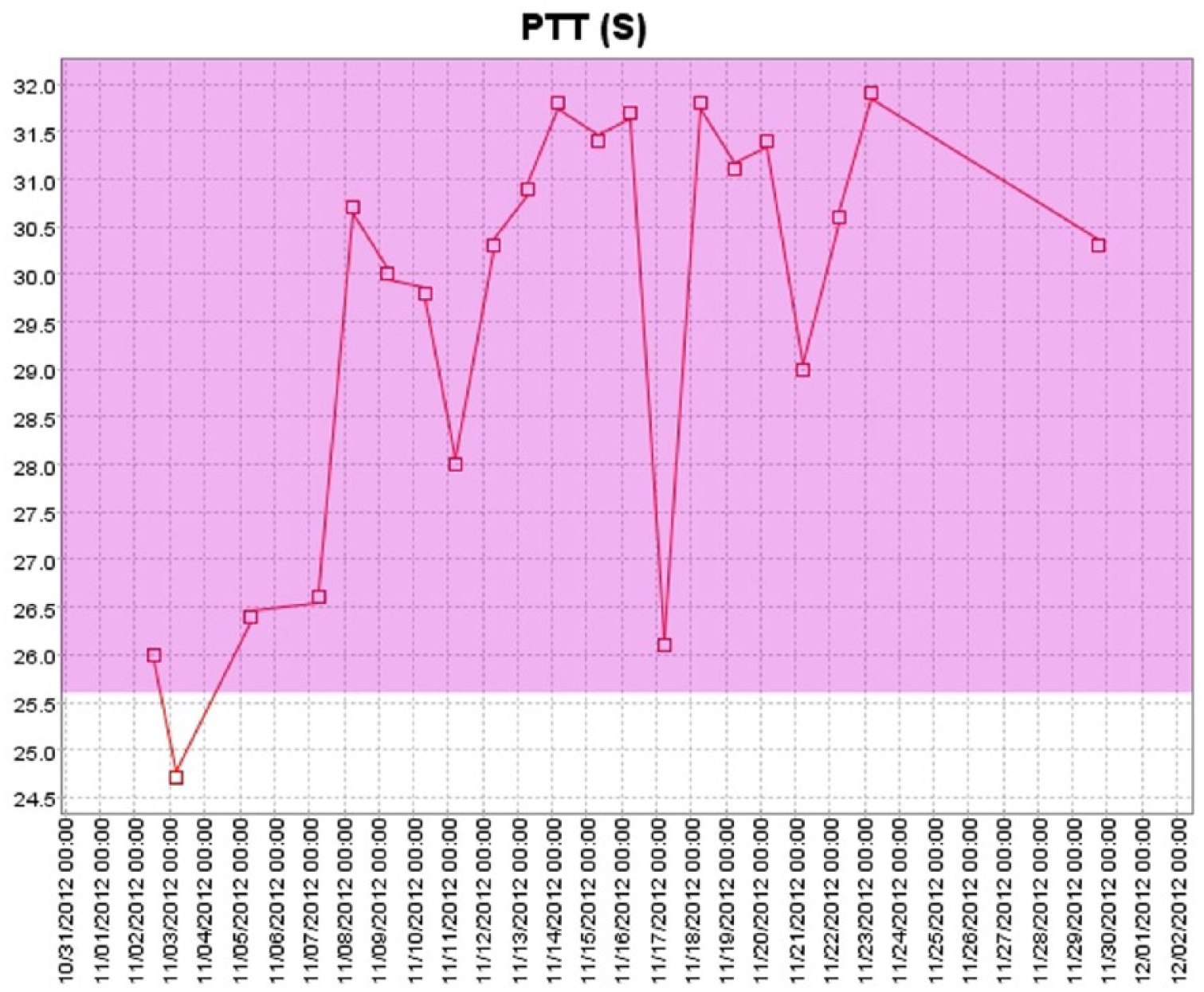
Changes in partial thromboplastin time (PTT) following first dose of FOLFOX.

Changes in international normalized ratio (INR) following first dose of FOLFOX.
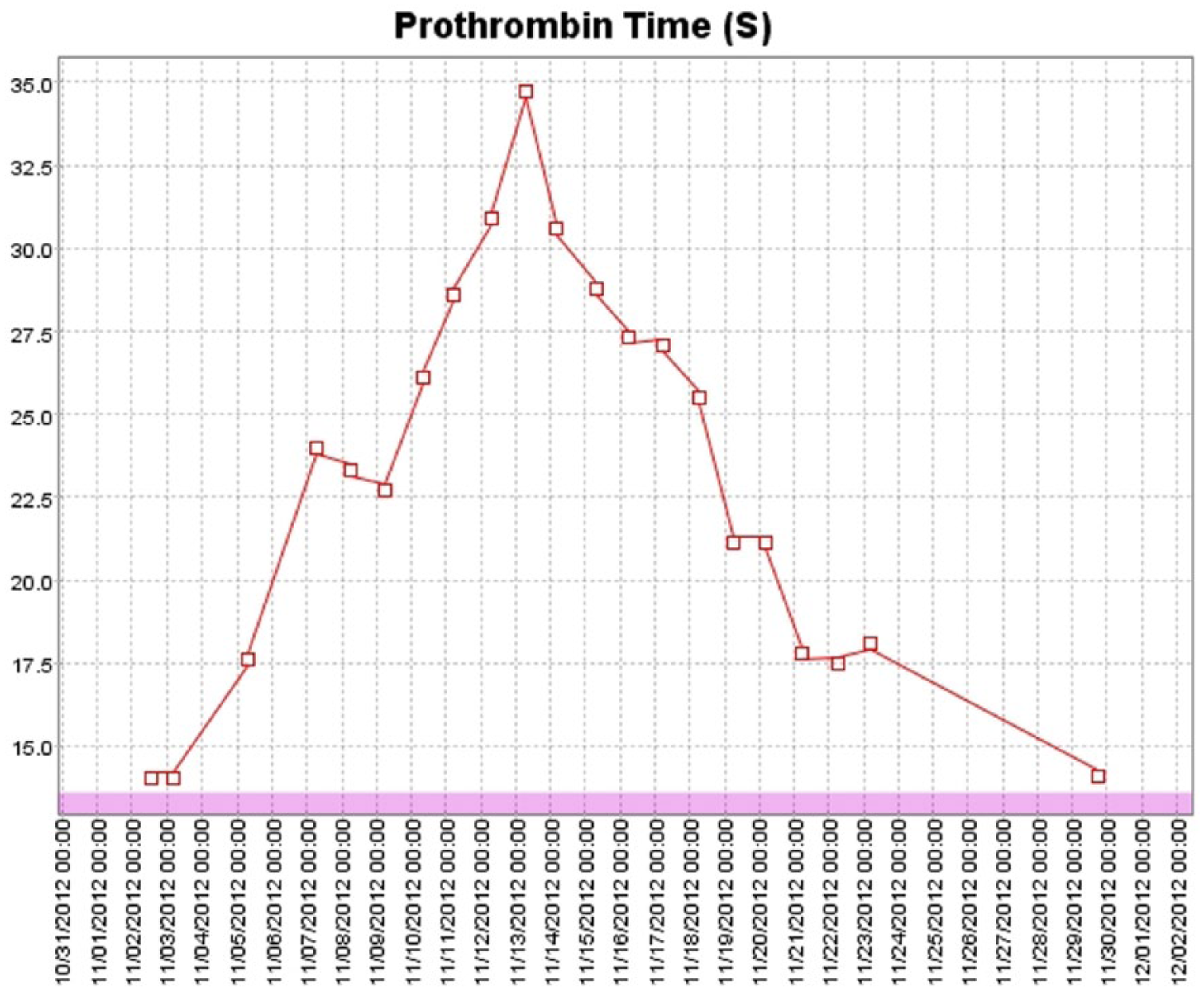
Changes in prothrombin time (PT) following first dose of FOLFOX.
Coagulation parameters following FOLFOX.

Vitamin K administered.
H, high; L, low; INR, international normalized ratio; PTT, partial thromboplastin time; PT, prothrombin time; F, factor.
Therefore, the patient was diagnosed with acquired isolated FVII deficiency. Review of the medical records showed normal range of both PT and INR both at the time of colonoscopy, the day before the placement of porta-catheter. Further review of family history revealed no history of hypercoagulable disease. The patient was not on warfarin and liver function tests were within normal range. The patient and her family refused surgery and she was placed in a hospice. She ultimately died 28 days later. No autopsy was performed.
As published earlier, DPYD/5FU pathway genotyping via matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry was performed at Dr Diasio’s laboratory by the methods described previously [Kinhult et al. 2003]. Within DPYDa agene, both rs115232898 (Y186C) and rs1801265 (C29R) were detected in the heterozygous state. The patient lacked any additional toxic effect-associated variations in the DPYD gene or the thymidylate synthase (TYMS) gene or promoter.
Discussion
5-FU or the FOLFOX regimen has not previously been associated with FVII deficiency to date. In addition, no previous reports exist about coagulopathies associated with DPD deficiency. Our patient constitutes the first report of FVII deficiency developing in a patient with DPD deficiency after receiving 5-FU. Kuzel and colleagues prospectively measured the assays for FPA (fibrinopeptide A), protein C, and protein S pre-infusion, 24 hours into the infusion, and post-infusion of 5-FU administered as a continuous infusion (CIV) at 1 g/m2/day over a 4- or 5-day period in ten patients (six with head and neck cancer and four with gastrointestinal malignancies) [Kuzel et al. 1990], [Kinhult et al. 2001]. They found an increase in FPA levels significantly during the CIV of 5-FU, which returned to the baseline at the end of 5 days of CIV. A significant decrease in protein C activity as compared with protein C antigen levels was also found. In an animal study, Kinhult and colleagues evaluated the effects of 5-FU, dalteparin, and the combination of these two substances on the vascular endothelium with scanning electron microscopy 1, 3, 7, 14, and 30 days after treatment and compared with a control group [Kinhult et al. 2001]. 5-FU-treated animals showed a severely damaged endothelium, often leading to intima disruption and denudation of underlying structures, with accompanying platelet accumulation and fibrin formation. The most extensive damage was observed on day 3 after treatment. The cytotoxic effect of 5-FU was partly reversible. The combination of 5-FU and dalteparin gave lower scores on day 3 because of less evidence of thrombotic events. The study suggested that antithrombotic treatment with dalteparin could protect against the thrombogenic effect of 5-FU, secondary to its direct toxic effect on the vascular endothelium [Trousseau, 1865]. The same investigators [Kinhult et al. 2003], in another experiment, treated five groups of rabbits (n = 75) with (1) 5-FU, (2) saline, (3) high-dose probucol and saline, (4) high-dose probucol and 5-FU, (5) low-dose probucol and 5-FU. Damage to the arterial endothelium was evaluated by scanning electron microscopy. Damage to the endothelium in 5-FU + probucol-treated animals was minimal and comparable to that of the control group. Intima disruption or thrombus formation was seen with 5-FU only. Further review of literature revealed a recent paper by Jenson and colleagues [Jensen and Sørensen, 2012]. They prospectively measured levels of plasma von Willebrand factor (vWf), urine albumin-to-creatinine ratio (UACR), coagulation factor II + VII + X, and fibrin D-dimer were serially assessed before, during, and after with 5-FU, folinic acid, and oxaliplatin. They found that vWf level increased, UACR increased, the coagulation factor II + VII + X activity decreased and the fibrin D-dimer level increased at baseline and during chemotherapy, respectively. In addition, they did not conclude any significance of these plasma biomarker changes to cardiovascular morbidity or its risk factors.
FVII is a vitamin-K-dependent enzyme of the serine protease class. When activated, FVII binds to tissue factor (TF). The TF–VII(a) complex activates both factors IX and X in the presence of phospholipid and calcium, and catalyzes the auto-activation of more FVII. The TF–VII(a) pathway is regarded as a dominant hemostatic mechanism in vivo [Saif et al. 2014]. The published incidence of the inherited form of Factor VII deficiency is estimated at 1 in 500,000. FVII deficiency is inherited in an autosomal recessive fashion caused by a mutation of FVII gene, located on chromosome 13, one of the 22 pairs of autosomal chromosomes. It affects males and females in equal number. A FVII-deficient person can be homozygous, double heterozygous, or heterozygous for the mutations in the FVII gene. Isolated acquired FVII deficiency in the absence of vitamin K deficiency, coumadin therapy, synthetic liver dysfunction, or overt DIC (disseminated intravascular coagulopathy) has been reported anecdotally. Other conditions include cancer, aplastic anemia, and antiphospholipid antibodies [Semeraro and Colucci, 1997; de Raucourt et al. 1994; Fischer et al. 1985; Weisdorf et al. 1989; Kimka et al. 1989]. In stem cell transplant (SCT) patients; a reduction in FVII levels has also been described, particularly in the first 2 weeks following SCT [Delmer et al. 1989; Bazzarbachi et al. 1993; Collins et al. 1994; Vannuchi et al. 1994]. In patients with less than 1% FVII activity, they present symptoms similar to hemophilia. Individuals with severe factor VII deficiency are prone to joint bleeds. In addition to spontaneous nosebleeds, GI bleeding (stomach, intestines) and urinary tract, head bleeds, muscle bleeds, and severe menorrhagia. Diagnosis is made through determination of activated partial thromboplastin time (aPTT) test, PT test and thrombin time (TT) test. Diagnosis can be confirmed with a FVII assay.
Our patient was found to have elevated FVIII. Battistelli and colleagues [Battistelli et al. 2008] investigated plasma concentrations of several coagulation factors and C4b-binding protein (C4BP) in 73 patients with nonmetastatic colorectal cancer (CRC; 48 colon and 25 rectum) and in 67 matched control subjects. Mean plasma concentrations of fibrinogen, FVIII, FIX, FV and C4BP were significantly higher in patients with CRC than in control subjects, while FVII and FXII levels were decreased significantly. Several correlations were found between the increased coagulation factors and C4BP concentrations, while FVII was highly correlated with FXII. It is possible that elevated FVIII in our patient along with elevated fibrinogen might be represent acute phase reactants.
Our patient did show improvement in INR following vitamin K administration but did not normalize. Previously, investigators published a retrospective series of eight patients who developed low plasma FVII level with normal levels of other coagulation factors following myelo-ablative chemotherapy and SCT [Toor et al. 2002]. In their patients, two patients were refractory to parenteral vitamin K supplementation and massive fresh frozen plasma (FFP) infusion and one patient was refractory to plasma exchange with FFP replacement. Another patient’s PT normalized upon entering remission. It has been suggested that a falling FVII level following SCT may be a sign of veno-occlusive disease of the liver (VOD) [Scrobohaci et al. 1992]. In the series referenced, three of the eight patients developed clinical findings of VOD. FVII levels were noted to be low when compared with FII and FX in the presence of chronic liver disease [Park et al. 1997]. In this condition, FVII deficiency is usually associated with deficiencies of other factors such as FXII or protein C.
The real pathogenesis leading to the idiopathic, acquired (nongenetic) deficiency of FVII is not known. Possible explanations may include development of an inhibitory or neutralizing antibody to FVII, development of an associated autoantibody to FVII such as a lupus inhibitor, or an autoimmune mechanism [Scrobohaci et al. 1992; Park et al. 1997; Okajima and Ishii, 1999; Greeno et al. 1995]. An acquired form of FVII deficiency has also been reported in patients with sepsis, postulated to be caused by cleavage of FVII by granulocyte-derived proteases [Biron et al. 1997]. Akin to this, a drop in FVIIc and FVIIa levels has been observed in normal volunteers study after administration of endotoxin [Li et al. 1996]. Another interesting explanation hypothesized by Drake and colleagues is the enhanced binding of FVII to TF. It is possible that high-dose chemotherapy might induce a state of increased vascular permeability, leading to an increased access of intravascular FVII to a large pool of extravascular TF, sequestering FVII in the extra vascular compartment [Drake et al. 1989]. The DPD deficiency syndrome manifests as exaggerated 5-FU toxicities including mucositis, hair loss, diarrhea, neutropenia, skin rash, and neurologic toxicities [Saif et al. 2007]. Complete DPD deficiency is extremely rare in the general population. About 3–5% of cancer patients are considered partially DPD deficient. In contrast to the incidence of approximately 3–5% in the White population, we recently reported that the incidence of DPD deficiency is slightly higher in the African-American population (8%) [Mattison et al. 2006] and our patient was also an African-American woman. FVII abnormalities or other such hemostatic abnormalities have not been reported to the best of our knowledge and there is no evidence linked to DPYD on chromosome 1p21.
In July 2005, the FDA approved recombinant FVIIa (NovoSeven) to treat bleeding episodes in patients with FVII deficiency. It is generally used for people with hemophilia (with FVIII or FIX deficiency) who have developed inhibitors against replacement coagulation factor [Hedner, 1998; White et al. 1999]. Reports of its use in the setting of uncontrollable hemorrhage are also scarce and carry the risk of arterial thrombosis. Further studies failed to show benefit and, therefore, are not used outside a clinical investigative setting. Prothrombin complex concentrates (PCCs) may offer some benefit but the amount of FVII contained in these products is variable. In addition, FFP can also be used. Our patient did not manifest any major bleeding and did response to vitamin K to improve (not normalize) INR.
In summary, we have described an uncommon form of acquired coagulopathy in the form of FVII deficiency arising in a patient following FOLFOX administration. Previous reports identified FVII deficiency in patients following SCT. DPD deficiency has been associated with severe 5-FU toxicity, but coagulopathy is not a common presentation. Further studies are needed to clearly elucidate the mechanism of this coagulopathy and whether patients with associated bleeding might be candidates for therapy with novel hemostatic agents.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there are no conflicts of interest.