
Abstract
Pancreatic neuroendocrine tumors (pNETs) are relatively rare malignancies. With secretory tumors such as insulinomas, vasoactive intestinal peptideomas, and gastrinomas, the hormone produced causes the symptom complex (e.g. hypoglycemia, peptic ulcer disease). With nonsecretory NETs, the clinical condition is determined by tumoral growth and metastasis. The course of metastatic pNETs may be indolent for several years but progression is often more rapid at later stages, leading to significant disability and a markedly negative impact on quality of life. Until recently, there were few effective systemic treatments for pNETs. Standard chemotherapy produces limited responses and has considerable toxicity. Somatostatin analogues control symptoms in some types of pNETs, but have not yet demonstrated antitumor activity. The recent introduction of targeted therapies, including the tyrosine kinase inhibitor sunitinib and the mammalian target of rapamycin inhibitor everolimus, yielded new opportunities for patients with advanced/metastatic pNETs. These drugs, which target key pathways in tumor proliferation and angiogenesis, provided clear clinical benefits in phase III clinical trials, including delayed tumor progression. The pivotal sunitinib phase III trial was discontinued prematurely due to higher rates of death and serious adverse events with placebo and greater progression-free survival (PFS) with sunitinib. In this trial, sunitinib demonstrated encouraging long-term responses as well as PFS and overall survival benefits, and an acceptable safety profile that allowed patients to preserve their quality of life. In every patient subgroup, including secretory and nonsecretory tumors, the hazard ratio for progression or death favored sunitinib. Circulating biomarkers are being investigated for the prediction and monitoring of responses to sunitinib. Although not fully evaluated in pNETs, biomarkers associated with response to sunitinib in several tumor types include soluble vascular endothelial growth factor receptor 2 and 3, interleukin 8, and stromal cell-derived factor 1α. Based on recent data, treatment algorithms have been updated for advanced and metastatic pNETs.
Introduction
Pancreatic neuroendocrine tumors (pNETs) are a rare subset of neuroendocrine tumors (NETs) originating from hormone-producing islet cells. They represent 1.3% of all pancreatic neoplasms [Metz and Jensen, 2008; Liakakos and Roukos, 2011] and their annual US incidence is estimated to be 0.32/100,000 [Yao et al. 2008a]. The pNET category encompasses various malignancies, including insulinomas, gastrinomas, and vasoactive intestinal peptideomas (VIPomas) (Table 1), with the symptoms and clinical course depending on the specific hormones produced (e.g. insulin, gastrin). The tumors are categorized as functional or nonfunctional based on hormone production, biological effects, and symptoms. Approximately 10–30% of pNETs are functional [Liakakos and Roukos, 2011]. They are also classified by degree of differentiation, with well differentiated tumors (grade 1 and 2) generally considered low grade and poorly differentiated tumors considered high grade, for which management may differ.
Recognized functional pancreatic neuroendocrine tumors and their characteristics.

Adapted from Milan and Yeo [2012] with permission from Lippincott Williams & Wilkins, Inc. and Ardill and O’Dorisio [2010] with permission from Elsevier Inc.
CgA is raised only in metastatic tumors.
CgA, chromogranin A; CgB, chromogranin B; PP, pancreatic polypeptide; VIP, vasoactive intestinal peptide; WDHA, watery diarrhea, hypokalemia, and achlorhydria.
Diagnosis of pNETs often occurs at an advanced stage because the symptoms are nonspecific and the disease course tends to be indolent [Vinik and Gonzales, 2011]. Even at an advanced stage, the tumors are slow to progress, with rates of 5-year survival in advanced disease estimated at 30–50% [Lepage et al. 2004; Panzuto et al. 2005; Yao et al. 2008a]. Localized disease can be cured surgically. However, most patients are not candidates for surgery at diagnosis due to nonresectable liver metastasis. In advanced, metastatic disease, debulking surgery may be useful and when metastases are limited to the liver, palliative benefits may result from locoregional treatments, including chemoembolization, radiofrequency ablation, and percutaneous ethanol injection [Delbaldo et al. 2012].
Until recently, no safe and effective systemic treatment was available for advanced pNETs. Conventional chemotherapy consisting of streptozocin with or without doxorubicin was associated with only modest response and considerable toxicity. For functional tumors, somatostatin analogues (SSAs) provide symptomatic relief but have limited antitumor activity [Liakakos and Roukos, 2011; Delbaldo et al. 2012]. Newly developed targeted treatments, such as sunitinib malate (SUTENT; Pfizer Inc., New York, NY, USA) and everolimus (AFINITOR; Novartis Pharmaceuticals, East Hanover, NJ, USA), have changed treatment practices for advanced, metastatic pNETs. These new drugs act by targeting key pathways involved in tumor proliferation and angiogenesis, and have demonstrated clear clinical benefits in phase III trials, including prolonged progression-free survival (PFS).
This review presents the rationale for targeted therapy in pNETs and provides an overview of clinical evidence and experience with sunitinib, including the use of quality of life (QOL) measures in clinical evaluation and the role of biomarkers in monitoring disease progression and response to treatment.
Rationale for sunitinib: cell signaling and angiogenesis
Somatostatin and somatostatin receptors as well as tyrosine kinase receptors [e.g. insulin-like growth factor I receptor (IGF1-R)] play major roles in controlling cell proliferation in NETs [Faivre et al. 2010]. Endothelial cells and pericytes are involved in tumor angiogenesis, with vascular endothelial growth factor (VEGF) and the VEGF receptor (VEGFR) governing endothelial cell survival [Inoue et al. 2002; Casanovas et al. 2005], and platelet-derived growth factor (PDGF) and the PDGF receptor (PDGFR) serving as important stimulating factors in pericyte functions [Fjallskog et al. 2003; Faivre et al. 2010]. Four major cell-signaling pathways drive cellular proliferation and survival for NET cell proliferation and angiogenesis: the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin (mTOR) pathway is involved in downstream VEGFR and PDGFR signaling, signal transduction of IGF1-R, and signal transduction induced by somatostatin receptors; the phospholipase C/protein kinase C pathway also plays a role in downstream VEGFR and PDGFR signaling; the RAS/RAF/mitogen-activated protein kinase pathway is involved in signal transduction of IGF1-R; and the Janus kinase/signal transducers and activators of transcription (STAT) pathway is involved in signal transduction induced by somatostatin receptors [Faivre et al. 2010].
Targeted therapies in pNET utilize two different mechanisms to inhibit tumor growth and survival (Figure 1). The tyrosine kinase inhibitor (TKI) sunitinib blocks activation of VEGFR and PDGFR, thereby inhibiting angiogenesis and inducing apoptosis in pericytes and endothelial cells. In contrast, the mTOR inhibitor everolimus binds specifically to mTOR, blocking signal transduction in tumor cells and cells that play a role in tumor angiogenesis [Faivre et al. 2010]. Targeted therapies that inhibit angiogenesis in pNETs appear to be effective because these tumors have well developed vasculature, their growth and survival are driven by VEGF, and angiogenesis plays a central role in tumor development. Angiogenesis appears to be greater in pNETs with a lower level of cell proliferation (measured by Ki67 expression) [Faivre et al. 2010]. Moreover, malignant pNETs demonstrate widespread expression of PDGFR-α and PDGFR-β, stem cell factor receptor (c-KIT), VEGFR-2 and VEGFR-3, as well as epidermal growth factor receptors [Couvelard et al. 2006; Faivre et al. 2010]. The central roles of VEGFR in endothelial cell survival and PDGFR in pericyte function make both receptors attractive therapeutic targets in pNETs.
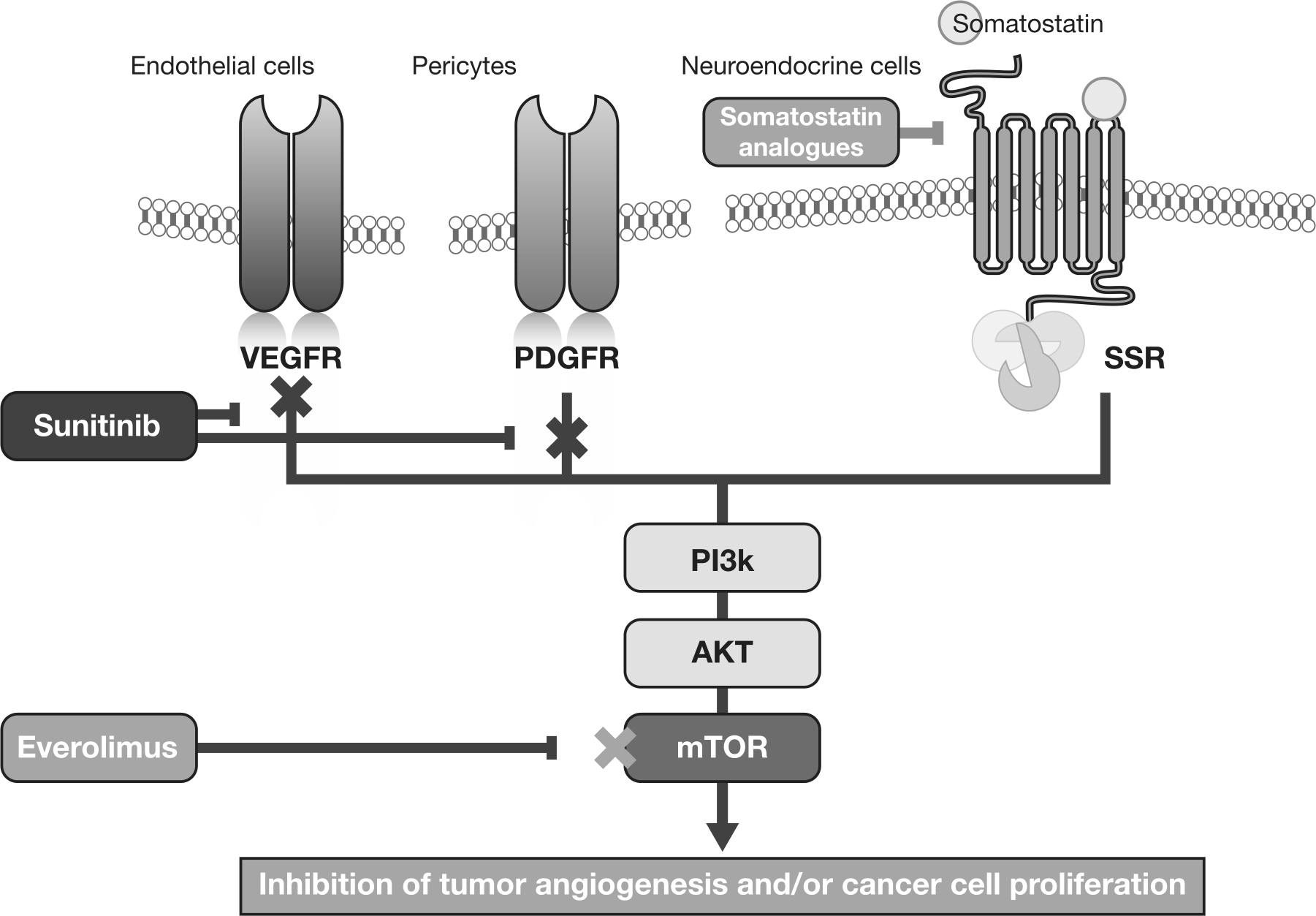
Mechanisms of action of targeted drugs in neuroendocrine tumors.
Sunitinib for pancreatic neuroendocrine tumors
Sunitinib malate is an oral multitargeted TKI with antiangiogenic and antitumor activity. It is a potent inhibitor of a range of receptor tyrosine kinases, including KIT, PDGFR-α and -β, and VEGFR-1, -2 and -3, Fms-like tyrosine kinase-3 receptor, and the receptor encoded by the ret proto-oncogene (RET) [Rubin et al. 2001; Pawson, 2002; Abrams et al. 2003; Mendel et al. 2003; Murray et al. 2003; O’Farrell et al. 2003; Kim et al. 2006]. Sunitinib has been approved multinationally for the treatment of imatinib-resistant/-intolerant gastrointestinal stromal tumor (GIST), advanced renal cell carcinoma (RCC), and progressive, well differentiated pNETs in patients with unresectable locally advanced or metastatic disease. In vitro, sunitinib inhibited growth in cell lines driven by VEGF, KIT, PDGF, RET, and colony-stimulating factor 1 receptor [Mendel et al. 2003; Faivre et al. 2007; Delbaldo et al. 2012]. In RIP1-Tag2 transgenic mouse models, it significantly reduced endothelial cell density and pericyte coverage of tumor vessels [Pietras and Hanahan, 2005; Delbaldo et al. 2012], and markedly reduced tumor blood flow and tumor volume [Olson et al. 2011].
Clinical experience with sunitinib in pancreatic neuroendocrine tumors
In a phase I dose-escalation trial, sunitinib showed strong antitumor activity in several solid tumor types, including RCC, GIST, and NET [Faivre et al. 2006]. The growth of these tumors is driven by tyrosine kinases, VEGF, KIT, and PDGF, and they are highly angiogenic and resistant to standard chemotherapy. Among the three patients who enrolled in phase I studies with NETs that had progressed after multiple cytotoxic agents, one achieved a partial response (PR) and the other two had sustained stable disease (SD) [Faivre et al. 2010]. Increased VEGF and decreased soluble VEGFR-2 (sVEGFR-2) levels demonstrated the targeted effects of sunitinib.
A phase II study was conducted to evaluate the safety and efficacy of sunitinib 50 mg/day, 4 weeks on and 2 weeks off (schedule 4/2) in advanced NETs [Kulke et al. 2008]. The study included 107 patients, including 66 with advanced pNETs and 41 with carcinoid tumors. Among patients with pNETs, sunitinib resulted in an objective response rate (ORR) of 16.7%, based on Response Evaluation Criteria in Solid Tumors (RECIST) criteria, with 56.1% achieving SD for over 6 months. The median time to tumor progression was 7.7 months.
Other trials have suggested that sunitinib may also be combined with transcatheter arterial embolization (TAE). Although TAE is an effective intervention in patients with NETs and liver metastases, embolization results in production and release of VEGF into the circulation. The safety and efficacy of sunitinib as an adjunct to TAE in advanced NETs with liver metastasis was evaluated in a phase II trial [Strosberg et al. 2012]. The study included 39 patients with NETs and liver metastases, including three patients with functional pNETs (one each of insulinoma, glucagonoma, and gastrinoma) who received sunitinib following TAE and were followed for a maximum of 12 months or until disease progression. The ORR was 72% [95% confidence interval (CI) 0.58–0.86] and median PFS was 15.2 months. The rates of overall survival (OS) at 1 and 4 years were 95% (95% CI 0.88–1.00) and 59% (95% CI 0.38–0.80) respectively. Serum VEGF showed a significant 34% increase following the TAE (p = 0.03).
The safety and efficacy of sunitinib given in a continuous daily dosing (CDD) schedule were evaluated in a phase II study of 12 Japanese patients with advanced, well differentiated pNETs [Okusaka et al. 2012]. The primary endpoint for the study was clinical benefit rate (CBR), defined as complete response (CR) + PR + SD for ≥24 weeks. Sunitinib was associated with potent antitumor activity, producing a CBR of 75% (95% CI 42.8–94.5), with 5 PRs and 4 SDs of at least 24 weeks. The ORR was 41.7% (95% CI 15.2–72.3) and the likelihood of PFS at 6 months was 91.7% (95% CI 53.9–98.8). Rates of adverse events (AEs) were consistent with the known safety profile for sunitinib. In the patient with gastrinoma who achieved PR, gastrin levels decreased by 93% and tumor bulk decreased by 45%.
Sunitinib 37.5 mg/day CDD was evaluated in a randomized, double-blind, placebo-controlled phase III study of 171 patients with well differentiated pNETs that had progressed within 12 months before study entry [Raymond et al. 2011]. The study was discontinued early due to higher rates of serious AEs and death in the placebo group and greater PFS in the sunitinib group. Median PFS for sunitinib was 11.4 months versus 5.5 months for placebo [hazard ratio (HR) for progression or death 0.42; 95% CI 0.26–0.66; p < 0.001). A subsequent reanalysis suggested possible differences in PFS outcomes, depending upon the observer. The ORR for sunitinib was 9.3% versus 0% for placebo (p = 0.007). This ORR did not include several patients who achieved unconfirmed PRs, due in part to the premature termination of the study, and also to the insufficiency of RECIST to accurately measure the antitumor effects of sunitinib. As a measure of response, RECIST is based exclusively on changes in tumor size. However, VEGF-inhibitor therapy can result in tumor necrosis in the interior of the lesion that appears as hypodensity on computed tomography (CT), with no accompanying changes in tumor size. Therefore, measurements of tumor effects using RECIST in the present study and others may have underestimated responses [Faivre et al. 2012].
At the data cutoff, nine deaths (10%) had occurred in the sunitinib group versus 21 (25%) in the placebo group (HR for death 0.41; 95% CI 0.19–0.89; p = 0.02). A Cox proportional-hazards model was used to assess the influence of patient and tumor characteristics on PFS. For each patient subgroup, the hazard ratio for progression or death favored sunitinib over placebo. Among patients with a Ki67 index of up to 5%, sunitinib clearly prolonged PFS versus placebo, and showed a trend toward a benefit among patients with a Ki67 index of over 5%. Sunitinib had a manageable safety profile, consistent with results from trials in RCC and GIST. Patient QOL was not adversely affected by sunitinib.
A series of studies and case reports have examined the use of sunitinib in patients with rare pNET subtypes. A small pilot study evaluated its safety and efficacy in 15 patients with Von Hippel–Lindau (VHL) disease, including seven patients with pNETs [Jonasch et al. 2011]. Patients received four cycles of sunitinib 50 mg/day (schedule 4/2). The overall safety profile was acceptable; five patients experienced grade 3 fatigue and 10 patients required dose reductions. All five evaluable patients with pNETs achieved SD, determined using modified RECIST criteria, with tumor shrinkage evident in all patients.
Bourcier and colleagues reported the successful use of sunitinib in two patients with pNET subtypes [Bourcier and Vinik, 2013]. The first was a 12-year-old boy with metastatic VIPoma who was 5 months post pancreatectomy and receiving octreotide 150 µg/h via pump with worsening symptoms. Sunitinib was titrated to 37.5 mg/day CDD, resulting in a confirmed PR based on RECIST. The PR correlated with decreases in biochemical markers, including VIP, pancreastatin, calcitonin, histamine, gastrin, and neurokinin. The young patient also exhibited a marked improvement in QOL. The second patient was a 70-year-old woman with metastatic paraganglioma/NET, who was given sunitinib 50 mg/day (schedule 4/2). She experienced a confirmed CR based on RECIST criteria, along with normalization of pancreastatin, metanephrines, and normetanephrines.
In a separate case report, sunitinib 37.5 mg/day and octreotide long-acting release (LAR) were administered to a 55-year-old woman with heavily pretreated advanced pNET [Grande et al. 2011]. After 3 months, there was a clear decrease in tumor density on CT (though no response according to RECIST criteria) and complete resolution of symptoms. The decrease in tumor density was accompanied by a decline in chromogranin A (CgA) levels. Similarly, a recent case report showed encouraging responses to sunitinib in a 28-year-old woman with pNET associated with VHL syndrome [Ali et al. 2012]. Treatment was initiated at 50 mg/day (schedule 4/2) after surgery for pancreatic and liver tumors, and then decreased to 25 mg/day due to toxicity. The patient achieved SD that has been maintained for 4 years. Sunitinib was also reported to significantly decrease tumor size, reduce CgA levels, and improve clinical status in a 37-year-old man with metastatic paraganglioma, using a dosing schedule of 50 mg/day (schedule 4/2), reduced to 25 mg/day due to toxicity [Cirillo, 2010].
Clinical considerations in treating pancreatic neuroendocrine tumors
The choice of treatment for pNETs includes medical, surgical, and multimodal approaches, depending on the location of the primary tumor and the presence of metastatic disease. Because pNETs are typically diagnosed at an advanced stage, the goals of treatment for surgically incurable disease center on symptom reduction, prolonged survival, and improved QOL. Measures to decrease tumor burden and associated symptoms include tumor debulking, selective ligation, and embolization with or without chemoembolization of the hepatic artery. In the case of locoregional disease, the goal of treatment is to stabilize the symptoms of functional tumors. Options include tumor enucleation using a minimally invasive technique and tumor resection with or without lymphadenectomy [Miljkovic et al. 2012].
For recurrent or metastatic disease, treatment options include targeted therapy with sunitinib or everolimus, both of which are considered first-line treatment in advanced disease. Cytotoxic chemotherapy is also indicated for advanced disease, including streptozotocin, adriamycin, 5-fluorouracil, dacarbazine, capecitabine, and temozolomide. The latter two agents are also considered first-line treatment options for advanced pNET. In a retrospective evaluation of 30 patients with metastatic endocrine carcinomas of the pancreas, first-line treatment with capecitabine plus temozolomide was associated with an ORR of 70% and a median PFS of 18 months. Nearly all patients (92%) were alive at 2 years. The toxicity of the combination was considerably less than that observed with streptozocin-based regimens [Strosberg et al. 2011].
Cytoreductive surgery may also be used in advanced disease if possible. Hepatic regional therapy options include hepatic artery embolization or chemoembolization and ablation (e.g. radiofrequency ablation, cryoablation, microwave ablation). Radioembolization with selective internal radiation therapy ameliorates symptoms, induces radiologic hepatic responses, and reduces CgA levels in patients with unresectable neuroendocrine liver metastases, with beneficial effects on long-term survival [King et al. 2008; Rajekar et al. 2011].
Approximately 80% of NETs express one or more types of somatostatin receptor; hence, the SSAs are a mainstay of treatment. They function as receptor agonists, reducing calcium channel activity and decreasing hormone secretion, and provide symptomatic relief and reduce biochemical marker levels in over 60% of patients with NETs. They also improve QOL with minimal AEs. Moreover, the placebo-controlled prospective randomized study on the antiproliferative efficacy of octreotide LAR in patients with metastatic neuroendocrine midgut tumors (PROMID) study showed that this SSA can even delay tumor growth [Rinke et al. 2009]. However, some patients with NETs become desensitized to SSA therapy over time [Miljkovic et al. 2012]. Radiolabeled or unlabeled SSAs such as octreotide, lanreotide, and pasireotide are indicated for symptom reduction in advanced disease. LAR formulations of octreotide and sandostatin allow monthly dosing [Miljkovic et al. 2012]. Responses to SSAs may vary by neuroendocrine tumor type. For example, gastrin and acid suppression can control gastrinomas [Yamaguchi et al. 2010], VIPomas [Song et al. 2009], and glucagonomas [Tomassetti et al. 1998] for many years, but insulinomas may improve or worsen and patients often develop hypoglycemia [Stehouwer et al. 1989]. Long-acting SSAs are incompletely effective at controlling diarrhea and indeed may cause diarrhea due to suppression of pancreatic enzymes [Vinik et al. 1982]. In addition, even with an initial response to long-acting SSAs, in about 40% of patients, their condition eventually fails to respond to this treatment and the breakthrough symptoms dictate the need for escape medication and combinations of short- and long-acting drugs [Delaunoit et al. 2008].
A variety of novel agents are currently being evaluated for treatment of pNETs (Table 2). These include a variety of targeted agents, novel antiangiogenesis agents, radiolabeled SSAs, and cytotoxic agents. The choice of targeted therapy for pNETs should be made on the basis of evidence from clinical trials, as well as patient-specific considerations. Phase II and III studies demonstrate that both sunitinib and everolimus provide clinical benefits, including increased PFS, in the treatment of advanced or metastatic pNETs (Table 3). Case reports and clinical trials underscore the importance of tailoring the dose and administration schedule to meet individual patient requirements and to minimize toxicity.
Overview of novel agents being evaluated for treatment of pancreatic neuroendocrine tumors.
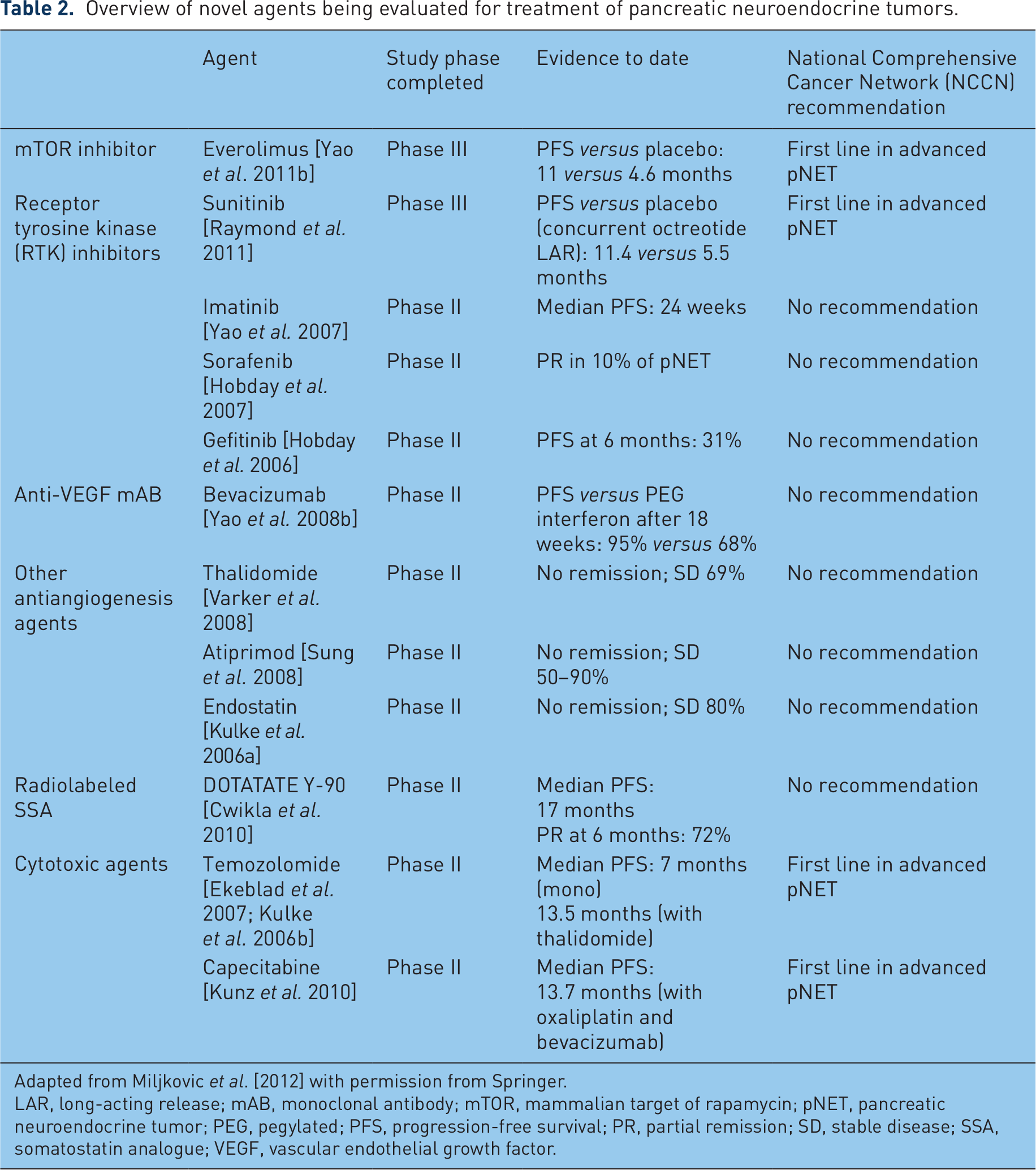
Adapted from Miljkovic et al. [2012] with permission from Springer.
LAR, long-acting release; mAB, monoclonal antibody; mTOR, mammalian target of rapamycin; pNET, pancreatic neuroendocrine tumor; PEG, pegylated; PFS, progression-free survival; PR, partial remission; SD, stable disease; SSA, somatostatin analogue; VEGF, vascular endothelial growth factor.
Results from phase II and III studies of sunitinib and everolimus in pancreatic neuroendocrine tumors.

Concomitant use of SSA in 27% of patients with pNET and 54% of patients with carcinoid tumours.
In patients with pNET.
Concomitant use of SSA in 26.7% of patients.
AE, adverse event; CDD, continuous daily dosing; CR, complete response; LAR, long-acting release; ORR, objective response rate; PD, progressive disease; PFS, progression-free survival; pNET, pancreatic neuroendocrine tumor; PR, partial response; RADIANT, RAD001 in Advanced Neuroendocrine Tumors; RCT, randomized, controlled trial; SD, stable disease; SSA, somatostatin analogue; TTP, time to progression.
In the absence of head-to-head comparisons, it remains unclear which of these targeted agents should be used first line and which reserved as second-line in pNETs (although everolimus can be considered first-line therapy in pancreatic malignant insulinoma because of the ability to control blood glucose). For individual patients, the safety profiles may provide a basis for choice. In a phase III study of everolimus, the most common AEs were stomatitis (64% overall, 7% grade 3–4), rash (49% overall, <1% grade 3–4), diarrhea (34% overall, 3% grade 3–4), fatigue (31% overall, 2% grade 3–4), and infections (23% overall, 2% grade 3–4). Noninfectious pneumonitis affected 17% of patients (2% grade 3–4), with one death caused by acute respiratory distress syndrome. Dose adjustments were required by 59% of patients [Yao et al. 2011b]. In the phase III study of sunitinib, the most common AEs were diarrhea (59% overall, 5% grade 3–4), nausea (45% overall, 1% grade 3–4), asthenia (34% overall, 5% grade 3–4), vomiting (34% overall, 0% grade 3–4), and fatigue (32% overall, 5% grade 3–4). One death was attributed to a sunitinib AE (cardiac failure). Dose interruptions were required in 30% of patients and dose reductions in 31%. However, serious AEs were less common with sunitinib than with placebo (26% versus 41%) [Raymond et al. 2011].
There may be differences in action among the compounds in the TKI class. Vandetanib has been approved by the US Food and Drug Administration for the treatment of late-stage medullary thyroid cancer (MTC) in adults ineligible for surgery [Thornton et al. 2012] and cabozantinib met its primary endpoint of PFS improvement in patients with MTC [Schöffski Elisei, Müller et al., 2012]. In our experience, sunitinib is not useful in MTC.
Biomarkers in pancreatic neuroendocrine tumors
The various subtypes of pNET are characterized by elevations in specific circulating hormonal and protein biomarkers (Table 1). These biomarkers are potentially useful in determining prognosis, monitoring disease progression, and assessing response to treatment. They offer the dual advantage of being noninvasive and inexpensive, but have limited utility for diagnosis due to a high rate of false-positive results. However, biomarker measurement does have some diagnostic value as a complement to radiologic imaging (CT, magnetic resonance imaging) and tissue pathology testing [Ardill and O’Dorisio, 2010].
A mutation in the multiple endocrine neoplasia (MEN1) gene is present in 15–20% of pNETs and is associated with poor prognosis. Patients with this mutation can have several tumors of different endocrine origins, and their neoplasms may even change tumor cell type. Patients with the MEN1 mutation should be regularly screened for abnormalities in insulin, gastrin, VIP, glucagon, somatostatin, pancreatic polypeptide, calcitonin, prolactin, and adrenocorticotropic hormone [Ardill and O’Dorisio, 2010].
Prognostic indicators can help to prevent overtreatment of indolent tumors, and aid in responding appropriately to rapidly growing tumors [Ardill and O’Dorisio, 2010]. Retrospective studies have identified several prognostic indicators shared by gastrointestinal NETs and pNETs, including Ki67 index, tumor volume, and metastatic spread [Shebani et al. 1999; Panzuto et al. 2005; Halfdanarson et al. 2008; Ahmed et al. 2009], and others specific to pNETs, such as degree of tumor differentiation [Panzuto et al. 2005].
Several circulating biomarkers have also been shown to have prognostic value in pNETs. For example, elevated baseline CgA and neuron-specific enolase (NSE) levels correlate with decreased PFS and OS [Yao et al. 2011a] and elevated baseline 5-hydroxy indole acetic acid correlates with increased PFS in advanced pNETs [Baudin et al. 2011]. In the phase II study of sunitinib in patients with pNETs or carcinoid, baseline interleukin 8 levels were significantly lower in those who achieved SD for over 6 months than in those who did not (p = 0.009) [Bello et al. 2006]. A later analysis from the same study found that elevated baseline sVEGFR-2 levels correlated significantly with improved OS in pNET (HR 0.22; 95% CI 0.06–0.78; p = 0.01) [Zurita et al., 2011]. No correlation was found between baseline sVEGFR-3 levels and outcomes. Elevated stromal cell-derived factor (SDF-1α) levels at baseline were found to predict a significantly shorter time to progression (HR 3.22; 95% CI 0.99–10.5; p = 0.05), PFS (HR 3.59; 95% CI 1.47–8.76; p = 0.005), and OS (HR 2.34; 95% CI 1.16–4.72; p = 0.02). Conversely, low baseline SDF-1α concentrations correlated with improved CBR, defined as objective response or SD of at least 6 months (p = 0.004).
Circulating cellular and protein biomarkers are also useful for monitoring the effects of treatment with targeted therapies, as well as disease recurrence (Table 4). Biomarkers are particularly useful as early indicators of progressive disease. For example, early decreases in CgA and NSE correlate with improved PFS in patients receiving targeted therapy [Baudin et al. 2011; Yao et al. 2011a], and in the phase II study of patients with pNETs or carcinoid tumors receiving sunitinib, reductions in sVEGFR-3 levels correlated with objective responses and improved PFS (p = 0.04). Among circulating cell types, CD14+ monocytes, particularly VEGFR-1+ and CXCR4+ monocytes, showed the largest decrease during treatment and may be useful as biomarkers of sunitinib response [Zurita et al. 2011]. Decreases in CgA levels have been associated with responses to sunitinib in individual patient cases [Cirillo, 2010; Grande et al. 2011] but larger studies have been inconclusive.
Soluble biomarkers and correlations with outcomes with targeted therapies in pancreatic neuroendocrine tumors.

Adapted from Mateo et al. [2012] with permission from Springer.
Patients randomized to everolimus + SSA or placebo + SSA.
5-HIAA, 5-hydroxy indole acetic acid; CBR, clinical benefit rate; CgA, chromogranin A; CI, confidence interval; HR, hazard ratio; IL-8, interleukin 8; MDACC, MD Anderson Cancer Center; NET, neuroendocrine tumor; NSE, neuron-specific enolase; OS, overall survival; PFS, progression-free survival; pNET, pancreatic neuroendocrine tumor; RADIANT, RAD001 in Advanced Neuroendocrine Tumors; SD, stable disease; SDF-1α, stromal cell-derived factor 1α; SSA, somatostatin analogue; sVEGFR, soluble VEGF receptor; VEGF, vascular endothelial growth factor.
Ki67, an indicator of cellular proliferation and mitotic activity, has also been proposed as a biomarker for response to targeted therapies such as sunitinib. Higher levels of angiogenesis are apparent in pNETs with low rates of proliferation, as measured by Ki67 expression. Therefore, elevated Ki67 may identify pNETs that will respond to antiangiogenic agents such as sunitinib [Faivre et al. 2010].
Health-related quality of life in patients with pancreatic neuroendocrine tumors
As novel treatments are prolonging survival, pNET has become a chronic condition for many patients, making QOL an increasingly important consideration. Patients with NETs have significantly worse QOL than the general population [Larsson et al. 2001; Frojd et al. 2007; Haugland et al. 2009; Pezzilli et al. 2009]. The symptoms of functioning pNETs, such as diarrhea, rash, and sweating, can have a profoundly negative impact on QOL, as can the toxicity associated with chemotherapy and radiation, and the emotional, social, and cognitive impact of the disease.
Measurement of QOL using questionnaires to assess patient-reported outcomes (PROs) in the setting of clinical trials is an important means of evaluating the benefit of treatments. PROs help to bridge the gap between the patient and physician, especially regarding sensitive personal issues related to treatment. Measurement of PROs in cancer trials has become a priority in the USA and Europe. Instruments include the National Cancer Institute’s Patient Reported Outcomes Measurement Information System and the European Organization for Research and Treatment of Cancer’s EORTC-QLQ-36 [Aaronson et al. 1993]. Questionnaires developed specifically to assess health-related QOL in NET include the Norfolk QOL-NET and the EORTC QLQ-C30 GI.NET-21 [Clauser et al. 2007; Vinik et al. 2011].
The Norfolk QOL-NET is a fully validated 72-item questionnaire, with excellent internal consistency [Vinik et al. 2009]. It covers seven domains: depression, flushing, respiratory, gastrointestinal, cardiovascular, physical functioning, and positive attitude. Measurements are related to a 4-week timeframe and capture 11 symptoms: frequency and severity of flushing, joint/bone pain, other pain, peripheral edema, wheezing, diarrhea/constipation, rash, cyanosis, telangiectasia, fatigue, and coughing. The questionnaire assesses the impact of these symptoms on daily activities, including work, family life, and pychosocial activities. The EORTC QLQ-C30 GI.NET-21, a 21-item questionnaire to supplement the QLQ-36 focusing on NETs of the gut, provides measurements using a 1-week timeframe, with the exception of measurements of sexual activity, which use a 4-week timeframe [Vinik et al. 2011]. It does not measure the frequency or severity of symptoms and does not address physical functioning. Additionally, it lacks items related to cardiovascular symptoms. It uses three defined multi-item symptom scales: endocrine symptoms, gastrointestinal symptoms, and treatment side effects, and has two single-item symptoms: bone/muscle pain, and worry about weight loss. It also uses two psychosocial scales (social functioning and disease-related worries) and includes two other single items (sexuality and communication). The questionnaire also includes items related to generic cancer concerns. A strong correlation has been demonstrated between Norfolk QOL-NET and the EORTC QLQ-C30 GI.NET-21. Either tool can be considered for use in the clinical trial setting and QOL as measured by both instruments has been correlated with Norfolk Carcinoid Symptom Score, tumor burden, and serotonin level (Table 5) [Vinik et al. 2011].
Comparison of Norfolk QOL-NET QOL total scores, Norfolk QOL-NET domains, tumor burden, biochemical markers, and Norfolk carcinoid symptom scores with EORTC QLQ-C30 GI.NET-21.

Reprinted from Vinik et al. [2011] with permission from Elsevier Inc.
CgA, chromogranin A EORTC, European Organization for Research and Treatment of Cancer; Norfolk QOL-NET, Norfolk quality of life – neuroendocrine tumor; QOL, quality of life.
The first randomized, controlled pNET trial to include QOL assessment was the phase III study of sunitinib [Vinik et al. 2010; Raymond et al. 2011]. The study used the EORTC QLQ-C30, a well validated QOL instrument suitable for the clinical trial setting, but not specific to pNET. Assessments of QOL measures were made every 4 weeks and the rate of compliance was over 80% (73 of 86 patients in the sunitinib group and 71 of 85 patients in the placebo group). No differences were found between sunitinib and placebo on the cognitive, emotional, physical, role, social functioning, and symptom scales, with the exception of diarrhea, which was significantly worse for patients on sunitinib. Post hoc analysis showed that the significant improvement in PFS with sunitinib was achieved without adversely affecting QOL. Sunitinib was also associated with delay in time to deterioration in QOL and emotional and physical functioning, which was dependent on PFS.
Conclusion
Targeted therapies have revolutionized treatment for advanced pNETs, improving efficacy and helping to maintain patient QOL. Clinical trials examining the use of targeted therapies in combination with other systemic treatments, as well as in adjuvant and neoadjuvant settings, may further extend the usefulness of these agents. Better understanding of biomarkers to measure treatment response and prognosis in pNETs may also allow more effective use of targeted and other therapies in the future.
Footnotes
Funding
Medical writing assistance was provided by Susanne Gilbert at ACUMED (New York, NY, USA) and was funded by Pfizer Inc.
Conflict of interest statement
Aaron Vinik has received institutional grants for research from Pfizer Inc., the manufacturers of sunitinib. Eric Raymond is a consultant to and has received institutional grants for research from Pfizer Inc., Novartis, and IPSEN.