
Abstract
Pancreatic ductal adenocarcinoma (PDA) is highly aggressive and has few treatment options. To personalize therapy, it is critical to delineate molecular subtypes and understand inter- and intra-tumoral heterogeneity. Germline testing for hereditary genetic abnormalities is recommended for all patients with PDA and somatic molecular testing is recommended for all patients with locally advanced or metastatic disease. KRAS mutations are present in 90% of PDA, while 10% are KRAS wild type and are potentially targetable with epidermal growth factor receptor blockade. KRASG12C inhibitors have shown activity in G12C-mutated cancers, and novel G12D and pan-RAS inhibitors are in clinical trials. DNA damage repair abnormalities, germline or somatic, occur in 5–10% of patients and are likely to benefit from DNA damaging agents and maintenance therapy with poly-ADP ribose polymerase inhibitors. Fewer than 1% of PDA harbor microsatellite instability high status and are susceptible to immune checkpoint blockade. Albeit very rare, occurring in <1% of patients with KRAS wild-type PDAs, BRAF V600E mutations, RET and NTRK fusions are targetable with cancer agnostic Food and Drug Administration-approved therapies. Genetic, epigenetic, and tumor microenvironment targets continue to be identified at an unprecedented pace, enabling PDA patients to be matched to targeted and immune therapeutics, including antibody–drug conjugates, and genetically engineered chimeric antigen receptor or T-cell receptor – T-cell therapies. In this review, we highlight clinically relevant molecular alterations and focus on targeted strategies that can improve patient outcomes through precision medicine.
Introduction
With a 5-year survival of only 11%, pancreatic ductal adenocarcinoma (PDA) is one of the deadliest tumors, highly resistant to chemotherapy, radiotherapy, and immunotherapy.1–5 Combination chemotherapy regimens, such as FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, oxaliplatin) and gemcitabine plus nab-paclitaxel (Gem-nabP), are standard first-line regimens in metastatic disease, with median survival less than 12 months.6–8 Neither regimen has so far been informed by biomarker status, although recent data suggest that low GATA-binding protein 6 expression by immunohistochemistry (IHC), correlating to a basal subtype PDA, may confer resistance to FOLFIRINOX, whereas no effect on survival was observed with Gem-nabP. 9
The promise of precision oncology has become a reality for certain malignancies, such as lung cancers and melanomas, and more recently, actionable targets have also been demonstrated in PDA. PDA is largely defined by core driver mutations in genes such as Kirsten ras (KRAS, 90%), tumor protein 53 (TP53, 64%), cyclin-dependent kinase inhibitor 2A (CDKN2A, 17%), and SMA and MAD-related protein 4 (SMAD4, 21%), but among them, only KRAS p.G12C mutations (1–3% of tumors) and TP53 p.Y220C mutations (0.64% of tumors) have been clinically targetable to date.10–13 Additional mutations in DNA damage repair (DDR) genes and chromatin-modifying genes are present at lower-level frequencies.14,15 KRAS wild-type status, present in 10% of PDA and up to 20% in younger patients, is an entity enriched with targetable alterations including microsatellite instability (MSI-high) and elevated tumor mutational burden (TMB high), ERBB2 amplification, BRAF mutations, as well as ALK, FGFR1-3, NRG1, NTRK1-3, RET, and ROS fusions.15,16
Besides somatic gene alterations, PDA is driven by germline genetic predisposition, with 3–8% of patients harboring deleterious germline variants in BRCA2/1, PALB2, ATM, CDKN2A, STK11, or mismatch repair (MMR) genes,17–20 with higher incidence in patients with a family history of PDA (10–13%),21–23 those from founder populations (up to 14.2% of Ashkenazi Jewish patients with a family history of breast and pancreatic cancer),24,25 and those with early onset PDA (28.6% of patients <50 years old at diagnosis). 26
A main research focus in PDA aims to connect molecular alterations with therapies targeting specific cellular pathways. Germline testing, comprehensive tumor next-generation sequencing, and liquid biopsies testing circulating cell-free DNA (cfDNA) reveal targets for therapeutic intervention. 27 In addition, immune-related biomarkers such as MSI-high, TMB-high, 28 inflamed T-cell signature profiles, 29 and tumor immune microenvironment phenotyping 30 have demonstrated predictive and/or prognostic implications. While PDA has been transcriptionally profiled into ‘basal-like’ and ‘classical’ associated with poor versus better prognosis, the value of selecting therapies based on these subtypes remains under investigation.14,31,32
Preliminary whole exome and RNA sequencing and clinical studies and registries support molecular testing in PDA patients and note the positive impact of personalized therapies on outcomes. Aguirre et al. identified therapeutically relevant genomic alterations in 48% of PDA patients, with 18% having pathogenic/likely pathogenic germline alterations. 33 The Pancreatic Cancer Action Network Know Your Tumor registry demonstrated that applying matched targeted therapies to molecular alterations doubled patient survival compared to standard of care chemotherapy. 34 The 2020 American Society of Clinical Oncology (ASCO) Guidelines 35 and National Comprehensive Cancer Network (NCCN) guidelines 6 have incorporated germline mutations testing for all newly diagnosed PDA, and somatic molecular testing from tumor biopsies or cfDNA (when tumor biopsy is not feasible) for all with locally advanced or metastatic disease.
Here we review the most significant actionable molecular alterations in PDA (Table 1, Figure 1) and summarize key clinical studies which evaluated the benefit of targeted therapies, including novel targets and studies in development (Tables 2 and 3).
Molecular testing in advanced PDA.

Ab, antibody; ADC, antibody–drug conjugate; DDR, DNA damage repair gene (e.g. BRCA1, BRCA2, PALB2); dMMR, deficient mismatch repair; HRD, homologous recombination DNA repair; MSI, microsatellite instability; NGS, next-generation sequencing; TKI, tyrosine kinase inhibitor; TMB, tumor mutational burden.
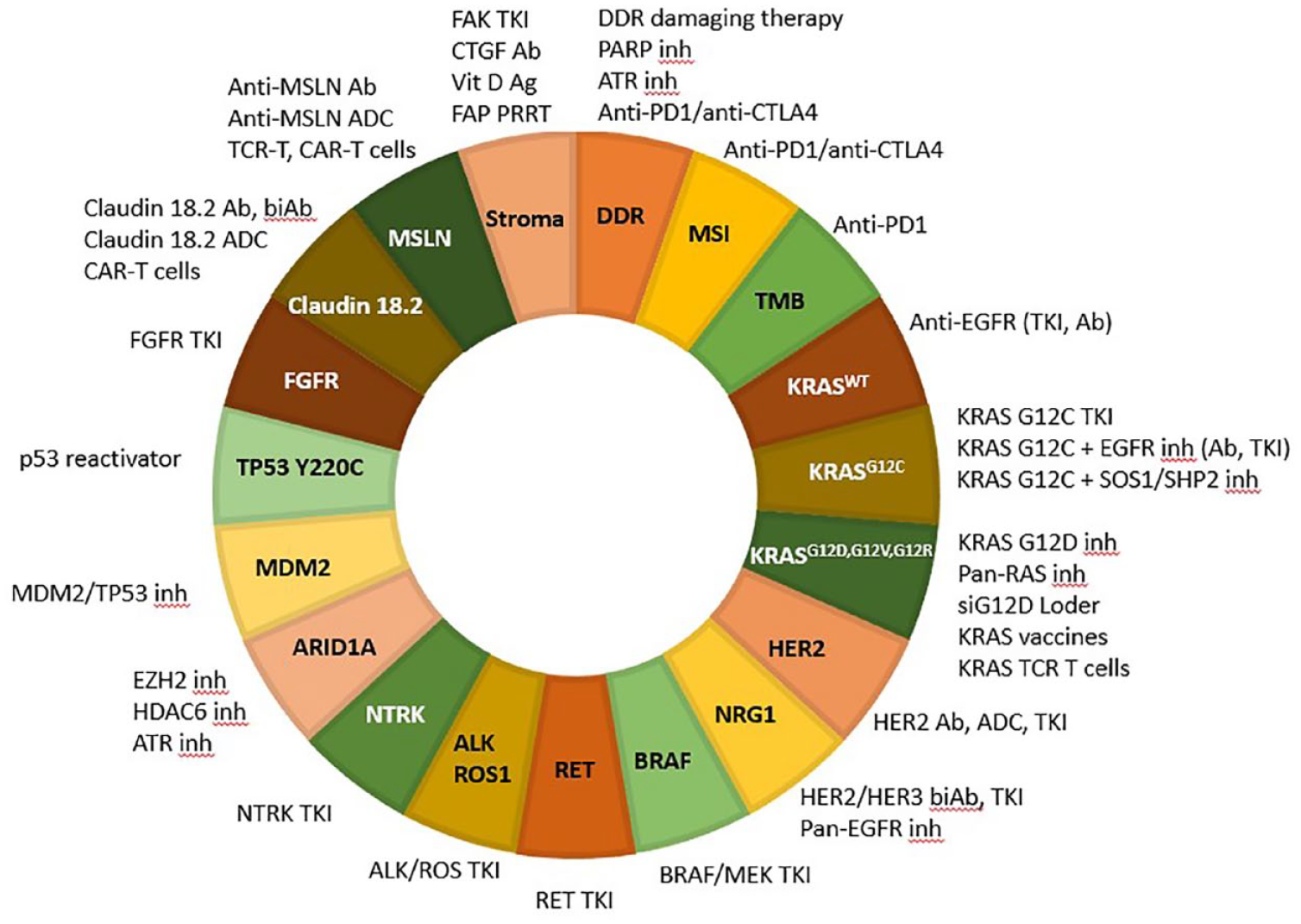
Key molecular targets and targeted therapies in PDA.
Active studies of targeted therapies in PDA.
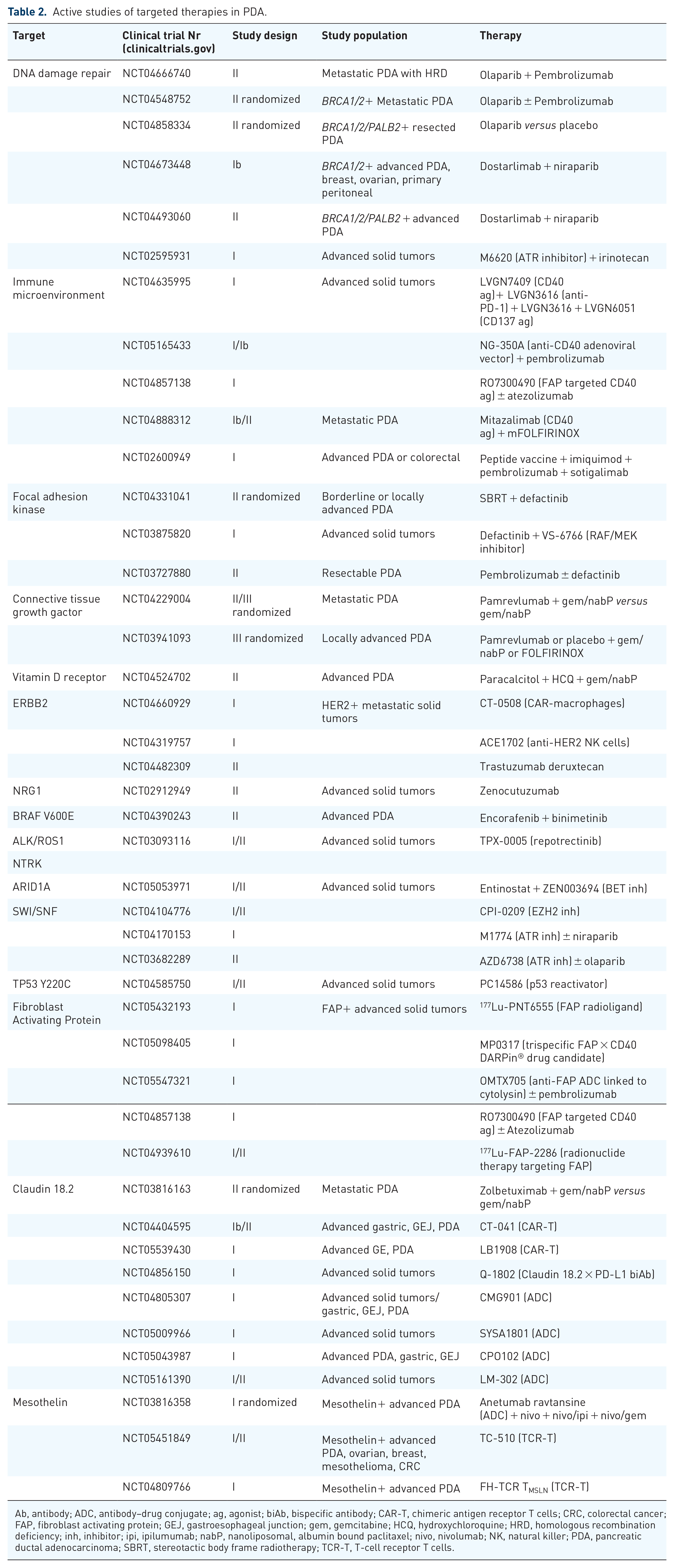
Ab, antibody; ADC, antibody–drug conjugate; ag, agonist; biAb, bispecific antibody; CAR-T, chimeric antigen receptor T cells; CRC, colorectal cancer; FAP, fibroblast activating protein; GEJ, gastroesophageal junction; gem, gemcitabine; HCQ, hydroxychloroquine; HRD, homologous recombination deficiency; inh, inhibitor; ipi, ipilumumab; nabP, nanoliposomal, albumin bound paclitaxel; nivo, nivolumab; NK, natural killer; PDA, pancreatic ductal adenocarcinoma; SBRT, stereotactic body frame radiotherapy; TCR-T, T-cell receptor T cells.
Targeting KRAS: active clinical trials.

DNA damage repair
Almost 20% of PDA harbor somatic or germline mutations in DDR genes such as BRCA1, BRCA2, PALB2, RAD51C, and RAD51D.36–38 Alterations in some DDR genes, especially biallelic inactivation of BRCA1 and BRCA2, can lead to a homologous recombination repair deficient (HRD) phenotype. HRD imparts susceptibility to irreversible DNA damage upon exposure to DNA damaging agents, including chemotherapy and radiotherapy, as well as poly-ADP ribose polymerase (PARP) inhibitors.38,39 While several DDR defects other than the core BRCA1/2/PALB2/RAD51 genes have been thought to impart HRD, recent data in PDA do not demonstrate an HRD phenotype or synthetic lethality with DNA damaging agents from non-core HRD genes.40–42 Moreover, not all BRCA1/2/PALB2 gene mutations confer loss of protein function, hence may not uniformly result in therapeutic benefit from targeted therapy with PARP inhibitors. Assessment of functional HRD is critical in identifying patients who benefit from PARP inhibitors. 43
Several assays such as Myriad’s MyChoice HRD CDx assay approved by the Food and Drug Administration (FDA) as a companion diagnostic for niraparib and olaparib in gynecologic malignancies detect BRCA1/2 mutations and compute a genomic instability score incorporating loss of heterozygosity, 44 large-scale transitions, 45 and telomeric allelic imbalances. 46 In addition, patterns of HRD single base substitution signatures, combined with indel micro-homology, and structural rearrangements created a weighted model called the HRDetect score, able to predict BRCA1/2 inactivation with a specificity of 98.7%. 47 The applicability of HRD scores to PDA is unknown.
Several clinical studies tested DNA damaging agents such as platinum chemotherapy and topoisomerase inhibitors, and PARP inhibitors in PDA including cancers those harboring BRCA1/2/PALB2 mutations. Olaparib, rucaparib, and veliparib conferred modest overall response rates (ORR) of 0–22% in previously treated germline BRCA1/2-mutated PDA.48-51 Combination strategies with chemotherapy have also been tested as first- and second-line treatment for metastatic PDA. First-line gemcitabine and cisplatin with or without veliparib in germline BRCA1/2-mutated PDA resulted in ORR of 65–74%, median progression-free survival (PFS) and overall survival (OS) of 10 and 19 months, respectively, with no benefit from veliparib. 52 The SWOG S1513 study tested second-line FOLFIRI with and without veliparib in unselected patients, but all underwent genomic sequencing with the BROCA-HR assay. 53 While veliparib did not improve survival among all patients (median OS 5.4 versus 6.5 months, and median PFS 2.1 versus 2.9 months with veliparib versus control), patients with core (BRCA1/2/PALB2) and non-core HRD alterations (e.g. ATM, ATR) versus wild-type HRD treated with FOLFIRI had higher median PFS (7.3 versus 2.5 months, p = 0.05) and OS (10 versus 6 months, p = 0.17).
Given overlapping myelosuppression and gastrointestinal toxicity between PARP inhibitors and chemotherapy hindering efficacy, maintenance strategies tested PARP inhibitors in patients with platinum-sensitive PDA [after response or stable disease (SD) from platinum chemotherapy]. The phase III POLO trial evaluated maintenance olaparib versus placebo in germline BRCA1/2-mutated PDA after at least 4 months of platinum-based chemotherapy without progression.54,55 Olaparib improved median PFS [7.4 versus 3.8 months; hazard ratio (HR): 0.53, p = 0.004], but not OS (19.2 versus 19.0 months; HR: 0.83; p = 0.3487). 56 The lack of significant OS improvement with olaparib in the POLO trial was likely due to the placebo-treated patients subsequent treatment with PARP inhibitors (27%) and with platinum or irinotecan-based chemotherapy upon progression. A similar benefit was demonstrated with maintenance rucaparib for platinum-sensitive germline or somatic BRCA1/2 or PALB2-mutated PDA: median PFS of 13 months (from starting chemotherapy) and OS of 23.5 months. 56
DDR defects increase genomic instability, neoantigenic load, and tumor immunogenicity. Furthermore, treatment with PARP inhibitors, by preventing DNA repair, upregulating programmed death-ligand 1 (PD-L1), and activating the stimulator of interferon genes pathway may synergize with immune checkpoint inhibitors (ICI).57,58 DDR deficiencies predict response to ICI in lung cancers, 59 but data in PDA are anecdotal. 60 In the CCTG PA.7 study, 61 16 patients harboring germline ATM mutations had higher OS with Gem-nabP plus anti-PD-L1/CTLA4 therapy with durvalumab/tremelimumab versus Gem-nabP alone (13.9 versus 4.9 months). 62 Ongoing clinical trials are testing combinations of maintenance olaparib with pembrolizumab for platinum-sensitive PDA: the POLAR study (NCT04666740) is testing olaparib plus pembrolizumab in cancers with core (BRCA1/2/PALB2) and non-core DDR mutations (ATM, BAP1, BARD1, BLM, BRIP1, CHEK2, FAM175A, FANCA, FANCC, NBN, RAD50, RAD51, RAD51C, RTEL1) and for platinum-sensitive DDR wild-type tumors, whereas the randomized SWOG S2001 study (NCT04548752) is testing olaparib plus pembrolizumab versus olaparib for germline BRCA1/2-mutated PDA. In ECOG-ACRIN 2192 (APOLLO) (NCT04858334), adjuvant olaparib versus placebo is being studied for germline or somatic BRCA1/2/PALB2-mutated PDA, after resection and completion of neoadjuvant/adjuvant chemotherapy. Reiss et al. recently reported on maintenance niraparib with the PD-1 inhibitor nivolumab or with the CTLA-4 inhibitor ipilimumab for platinum-sensitive PDA. The combination of PARP/CTLA4 versus PARP/PD-1 blockade conferred superior 6-month PFS: 59.6% versus 20.6%, 63 and increased ORR, median PFS and OS: 15.4% versus 7.7%, 8.1 versus 1.9 months, and 17.3 versus 13.2 months, respectively. Mutated DDR genes beyond the core BRCA1/2/PALB2 genes have been associated with benefit from platinum chemotherapy, but small patient numbers preclude definitive conclusions. 64 PARP inhibitors plus ICIs are also investigated for refractory PDA (NCT04673448, NCT04493060).
Despite initial benefit, resistance to PARP inhibitors eventually develops. 65 Several mechanisms of resistance lead to restoration of homologous recombination DNA repair and persistent replication forks. 66 Preclinical data suggest that PARP inhibitor-resistant BRCA1-deficient cells are increasingly dependent on ATR for survival. 67 Another strategy tackling PARP inhibitor resistance is with the PARP/WEE1 inhibitor combination which induces replication stress. 68
Data with PARP inhibitors in PDA with non-BRCA1/2/PALB2 DDR alterations showed low efficacy. 43 Germline and somatic ATM defects occur in 5–10% of PDA. 37 ATR inhibitors are active in ATM-deficient cancers,69–72 and given preclinical synergism with carboplatin and irinotecan, studies are evaluating these combinations (NCT02595931). No studies reported on PARP plus ATR inhibitors in PDA, but the phase II VIOLETTE trial in breast cancer did not show increased efficacy from this combination. 73
Novel research identified HRD and replication stress as broader targets, beyond BRCA1/2/PALB2, encompassing the functional relevance from other DDR alterations. 74 These provocative results highlight that molecular profiling may identify additional therapeutic vulnerabilities.
Microsatellite instability
MSI high/deficient DNA mismatch repair (dMMR) tumors may benefit from ICIs.75,76 The MMR system recognizes and repairs the erroneous insertion, deletion, and misincorporation of bases that arise during DNA replication and recombination. Tumors harboring MSI-H/dMMR can accumulate thousands of mutations and are characterized by a hypermutated genome, correlating with response to ICI. Testing for MSI-H/dMMR can be done using IHC and molecular tests including polymerase chain reaction-based MSI testing and novel next-generation sequencing (NGS) approaches. 77
The frequency of MSI-H/dMMR occurs in approximately 1% of PDA, either in the context of Lynch syndrome17,18,78 or as somatic mutations.79–81 In one systematic review, MSI-H/dMMR was strongly associated with medullary and mucinous/colloid histology as well as with wild-type KRAS and TP53 tumors. 82 Moreover, TMB is elevated [defined as ⩾10, and in some studies ⩾20 mutations/megabase (mut/Mb)] in the majority of MSI-H/dMMR PDA, representing another biomarker associated with benefit from anti-programmed cell death protein 1 (PD1)/PD-L1 agents. 83
The US FDA-approved pembrolizumab based on ORR of 40% among 149 MSI-H/dMMR cancers. 84 In the initial cohort of non-colorectal cancers, five of eight (62%) PDA patients responded. 76 The follow-up KEYNOTE-158 trial noted ORR of 34%, with a median PFS of 4 months and an OS of 23.5 months among pretreated MSI-H/dMMR solid tumors, but 22 PDA patients had ORR of 18.2%, and median PFS and OS of only 2 and 4 months, respectively. 85 These results emphasize the lower benefit from ICI for MSI-H PDA, likely the result of a profoundly immunosuppressive microenvironment.
Tumor mutational burden
Another biomarker with predictive benefit from anti-PD1 therapies is the TMB, commonly reported in comprehensive NGS assays.58,86,87 TMB is a numeric index of the estimated total number of mutations per coding area of the tumor genome. 88 High-TMB is associated with response to anti-PD-1 therapies.86,89 TMB is considered high if it exceeds a predetermined threshold, widely variable based on tumor type. 90 In a retrospective analysis of KEYNOTE-158, patients with TMB-high tumors, defined as a TMB ⩾ 10 mut/Mb, demonstrated a significantly higher ORR to pembrolizumab compared to tumors harboring TMB < 10 muts/Mb (29% versus 6%). 91 In contrast, Schrock et al. identified 37 mut/Mb as the optimal cutoff for colorectal cancers. 92 Little is known regarding the prevalence and the potential predictive role of TMB in PDA. In a systematic review, TMB-high defined as ⩾20 mut/Mb was present in 1.1% of PDA. 28 A significant portion (59.4%) of TMB-high cancers harbor MSI-H/dMMR, while MSS TMB-high tumors have BRCA2, BRAF, or POLE mutations. In this analysis, eight patients with TMB-high PDA received anti-PD1 therapy: two (also MSI-H/dMMR) had complete response (CR), five partial response (PR), and one SD lasting for 30 months. In a retrospective analysis of 3500 PDA samples, Singhi et al. considered TMB-high as ⩾20 mut/Mb, but only 0.5% of tumors met this criterion. 16 Confirming the need for a higher TMB, a recent analysis of 1678 MSS solid tumors including 26 PDA with TMB ⩾ 10 mut/Mb treated with pembrolizumab showed response in only one (4%) PDA. 93
TMB typically ranges between 1 and 3 mut/Mb in PDA, 93 and it does not appear to be a reliable predictor of benefit from ICI. Nonetheless, in the CCTG PA.7 study of Gem-nabP with and without durvalumab/tremelimumab, cfDNA analysis showed that in a small subset of patients (n = 8) with plasma TMB ⩾ 9 mut/Mb, patients had higher median OS with chemoimmunotherapy versus chemotherapy alone (14.6 versus 1.2 months).61,94 Recent reports indicate TMB ⩾ 10 mut/Mb to be more common in KRAS wild-type (4.5%) compared to KRAS-mutated PDA (1%), 15 correlating to higher incidence of MSI-high in this subtype. DDR alterations including biallelic mutations in BRCA1/2/PALB2, ATM, BARD1, BLM, CHEK2, RAD50, and RAD51C have higher TMB and genomic instability, 95 and these genomic alterations could represent biomarkers for combination ICIs, including anti-PD1, anti-CTLA4, anti-PD1/anti-CTLA4 inhibitors with and without DDR blockade.
NCCN guidelines recommend pembrolizumab for patients whose cancers have TMB ⩾ 10 mut/Mb and have failed prior therapies, or in the first-line setting for patients with poor performance status, not fit for conventional chemotherapy. 6
Tumor microenvironment
The tumor microenvironment (TME) in PDA consists of immune cells, cytokines, metabolites, fibroblasts, and desmoplastic stroma rich in hyaluronic acid (HA) and collagen. This multifaceted compartment is thought to be, in part, responsible for the resistance to most chemo and immunotherapies.95–98 The immunosuppressive TME is characterized by limited infiltration of CD8+ T cells and an abundance of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor-associated neutrophils, and regulatory T cells. In most patients, the TME prevents anticancer immunity and promotes carcinogenesis. However, a robust host immune response has been identified, with abundant CD8+ T-cell infiltrates and high number of neoantigens in long-term survivors after PDA surgery. 99
Single agent and combinations of ICI have been ineffective in advanced PDA, but many clinical trials testing new combinations are underway.100–102 The randomized phase Ib PRINCE trial evaluated Gem-nabP with nivolumab, with the CD40 agonistic monoclonal antibody sotigalimab (aimed to activate dendritic cells and repurpose immunosuppressive M2 to proinflammatory M1-TAMs), or with nivolumab plus sotigalimab. 103 While only the nivolumab/Gem-nabP arm improved 1-year OS (57.7% versus historical 35% control, p = 0.006), distinct immune signatures were associated with survival in each arm. A less suppressive TME and higher numbers of activated antigen-experienced circulating T cells at baseline predicted benefit from nivolumab/Gem-nabP, while greater intratumoral CD4+ T cells and circulating differentiated CD4+ T cells and antigen presenting cells predicted benefit from sotigalimab/Gem-nabP. 104
It has been well described that KRAS mutations negatively impact the TME in PDA. 105 Recently, differences were noted in the immune microenvironment of KRAS wild-type and KRAS-mutated PDA, with a larger proportion of infiltrating, active effector T cells, and fewer MDSCs in KRAS wild-type tumors, suggesting that this subtype may be more susceptible to targeting by ICI, and possibly accounting for improved survival. 15
The extracellular matrix promotes the immunosuppressive TME and impedes perfusion by compressing the tumor vasculature. Targeting stroma intends to improve systemic drug delivery and allow effector immune cell infiltration. Several stromal targeting strategies have been tested, most without the guide of a predictive biomarker. HA content has been thought to predict benefit from pegvorhyaluronidase alfa (PEGPH20) based on preclinical data, and due to encouraging results in combination with gemcitabine or with Gem-nabP for PDA with high HA content.106,107 Nevertheless, the phase III HALO-301 study in HA-high PDA demonstrated an equivalent median OS of 11 months with Gem-nabP with or without PEGPH20. 108 A possible explanation for the lack of benefit from PEGPH20 was that biomarker selection of the HA-high threshold as 50% HA expression of any intensity by an IHC assay was not adequate. Other stroma targeting strategies, albeit without biomarker selection, include the vitamin D receptor agonist paricalcitol with chemo- and immunotherapy, 109 focal adhesion kinase inhibition with defactinib, VS-6766 or IN10018 with chemotherapy, radiotherapy and/or ICI (NCT02758587, NCT02546531, NCT04331041), 110 and connective tissue growth factor (CTGF/CCN2) blockade with pamrevlumab (FG-3019) with chemotherapy and/or ICI (NCT04449004, NCT03941093, NCT03727880, NCT04331041) (Table 2). 111
Fibroblast activating protein (FAP) is a type II membrane bound glycoprotein which activates cancer-associated fibroblasts. 112 FAP is expressed on activated fibroblasts in tumors stroma. FAP targeting has previously been unsuccessful, but FAP imaging may select patients for FAP-targeted therapies. More recently, FAP has been identified as a potential target for peptide receptor radionuclide therapy. 113 Results with 90Y-labeled FAPI-46 radioligand therapy for refractory solid tumors with high FAP expression by PET/CT have been recently reported. 114 Among 119 screened tumors, 21 were eligible (3 PDA). ORR and disease control rate (DCR = CR + PR + SD) were 6% and 38%, respectively, but no response/SD occurred in PDA. Another phase I study is exploring [Lu-177]-PNT6555 in FAP-avid solid tumors as determined by the [Ga-68]-PNT6555 PET/CT (NCT05432193).
KRAS
Oncogenic KRAS mutations occur in >90% of PDA and are the hallmark of this disease. KRAS encodes a small GTPase, which oscillates between active (GTP-bound) and inactive (GDP-bound) state, and when active, signals to major downstream pathways: RAF/MAPK/ERK and PI3K/AKT/mTOR.115–118 KRAS mutations in codons G12, G13, and Q61 prevent GTPase-activating proteins from hydrolyzing GTP to inactive GDP. Signals from receptor tyrosine kinases (RTK) flow through adapter proteins to son of sevenless homolog 1 (SOS1), a key guanine exchange factor which promotes GDP exchange to GTP, and Src homology region 2-containing protein tyrosine phosphatase (SHP2). 117 PDA are enriched in KRASG12D (35.5%), KRASG12V (28.2%), and KRASG12R (15.9%) point mutations.12,15 Due to lack of accessible binding pockets, most efforts to target KRAS directly have been difficult. However, KRASG12C (1–3% of PDA) has recently emerged as an actionable target. 12 In addition, novel KRASG12D and pan-RAS inhibitors, as well as SOS1 and SHP2 inhibitors as monotherapy and in combinations, entered clinical trials (Table 3).
KRAS G12C
The mutant cysteine-12 is located next to a cryptic pocket of the switch II region in the inactive GDP-bound conformation of KRAS. Several small-molecule covalent inhibitors bind specifically and irreversibly to mutant cysteine and disrupt both switch I and switch II exchange factors, trapping mutant KRAS in an inactive GDP state. Sotorasib (AMG510) and adagrasib (MRTX849), among others, have been optimized for favorable pharmacokinetic (PK) properties including long half-life, extensive tissue distribution, dose-dependent PK, as well as central nervous system penetration. In the phase I/II KRSYTAL-1 study, adagrasib was evaluated in patients with advanced solid tumors harboring KRASG12C mutations, including 12 heavily pretreated PDA. 119 Among 10 evaluable patients, 5 responded (50%), DCR was 100%, and a median PFS was 6.6 months. Sotorasib was evaluated in the phase I/II CodeBreaK100 trial in advanced solid tumors harboring a KRASG12C mutation, including 38 PDA. 120 Most patients (79%) had ⩾ 2 prior lines of therapy. The ORR was 21.1%, DCR was 84.3%, and median PFS and OS were 4 months and 6.9 months, respectively.
While KRASG12C inhibitors are well-tolerated, the benefit is transient. Mechanisms of resistance include secondary KRAS mutations, alterations in cell cycle regulation, activating mutations in other RTK and downstream RAS-MAPK pathways, and emergence of new gene fusions. 121 The diversity of resistance mechanisms supports the development of combination regimens, including with agents targeting EGFR, SHP2, SOS1, MEK, CDK, mTOR, targetable fusions, and PD-1 inhibitors (Table 3).
KRAS non-G12C
Inhibitors targeting other KRAS variants, such as KRASG12D (MTRX1133, siG12D Loder), pan-RAS inhibitors (RMC6236, BI1701963), novel KRAS vaccines, and adoptive immunotherapies targeting various KRAS alleles are in development and expected to broaden efficacy in PDA patients (Table 3). Adoptive chimeric T-cell receptor (TCR) T cells therapy has been successfully used in a previously treated PDA patient with lung metastases who obtained a PR and a PFS of 6 months+. 122 This patient received autologous T cells genetically engineered to clonally express two allogeneic HLA-C*08:02-restricted TCRs targeting mutant KRAS G12D. Several TCR cell therapies targeting KRAS variants are being explored. Future trials are likely to include combination immunotherapy approaches and next-generation TCR-T cells with chimeric co-stimulatory molecules, such as activating receptors or ligands.
KRAS wild type
Approximately 10% of PDA and up to 20% of young-onset PDA (age <50 years) are KRAS wild type (KRAS WT ), with better prognosis and more therapeutic opportunities. 15 KRAS WT PDAs are enriched in BRAF, DDR, chromatin remodeling, cell cycle control gene mutations, FGFR2, ALK, RET, NTRK and NRG1 fusions, as well as FGF3, ERBB2, FGFR3, and MET amplifications, and are more likely to exhibit MSI-high and TMB-high status. In all, almost 30% of KRAS WT PDA have targetable alterations.
Several reports in the past decade suggested that KRAS WT PDA have a better prognosis overall, including when treated with anti-epidermal growth factor receptor (EGFR) therapies. In 2005, based on the phase III study NCIC CTG PA.3, the FDA approved the anti-EGFR tyrosine kinase inhibitor (TKI) erlotinib combined with gemcitabine in metastatic PDA due to a 18% improvement in median OS versus gemcitabine and placebo (6.2 versus 5.9 months, HR: 0.82; p = 0.03). 123 This study did not observe significant correlations with KRAS status or EGFR expression among 26% of cancers with available tumor samples for analysis, but OS was higher for KRAS WT PDA treated with gemcitabine/erlotinib versus gemcitabine/placebo (6.1 versus 4.5 months, HR: 0.66, p = 0.34), whereas KRAS-mutated (KRASMUT ) PDA derived no benefit (6.0 versus 7.4 months, HR: 1.07, p = 0.74). 124 Kim et al. noted a 4-month survival advantage for KRAS WT versus KRASMUT PDA treated with gemcitabine/erlotinib (OS: 9.7 versus 5.2 months, p = 0.002). 125 Similarly, a retrospective biomarker analysis in the phase III study AIO-PK0104 identified KRAS WT versus KRASMUT to confer superior OS (7.9 versus 5.7 months, HR: 1.68, p = 0.005) for patients treated with chemotherapy plus erlotinib. 126 Increased OS was also noted with the anti-EGFR antibody cetuximab plus gemcitabine/oxaliplatin among KRAS WT versus KRASMUT PDA (8.7 versus 5.4 months). 127
No prospective randomized phase III study evaluated anti-EGFR therapies versus placebo in KRAS WT PDA until the NOTABLE study was reported at ASCO 2022. 128 Nimotuzumab, a humanized IgG1 monoclonal antibody against the extracellular domain of EGFR, 129 showed encouraging efficacy with gemcitabine versus gemcitabine alone in a randomized phase II study (OS: 8.6 versus 6 months), and OS was significantly higher (11.6 versus 5.6 months) in KRAS WT PDA. 130 Based on these results, the phase III study NOTABLE evaluated nimotuzumab plus gemcitabine versus placebo plus gemcitabine for first-line treatment of KRAS WT advanced PDA. 128 Nimotuzumab significantly increased median OS (10.9 versus 8.5 months, HR 0.50, p = 0.024). This study should provide impetus for further exploration of anti-EGFR therapies in KRAS WT PDA in combination with contemporary multi-agent chemotherapy.
ERBB2/HER2
Human epidermal growth factor receptor 2 (HER2) overexpression occurs in 2–3% of PDA.131,132 HER2 and HER3 are obligate partners and together are implicated in the progression of multiple cancers. 133 Monoclonal antibodies that bind either directly to the extracellular domain of HER2 (e.g. trastuzumab), block the interaction of HER2 and HER3 (e.g. pertuzumab), or the antibody–drug conjugate (ADC) trastuzumab deruxtecan carrying a topoisomerase I inhibitor payload, are currently in clinical practice for breast and gastroesophageal cancers.134,135
Several studies tested anti-EGFR/HER2 TKIs afatinib or lapatinib in biomarker unselected PDA, without significant benefit.136–139 The MyPathway phase II basket study tested trastuzumab plus pertuzumab in 258 refractory cancers, including 10 PDA with HER2 overexpression by IHC, fluorescence in situ hybridization (FISH), or NGS. 140 ORR was 23.3%, higher in KRAS WT (26%) versus KRASMUT tumors (4%). ORR was 33% in KRAS WT PDA.
Neuregulin-1
Neuregulin-1 (NRG-1) is a ligand which binds primarily to ERBB3/HER3 and ERBB4/HER4, leading to hetero- or oligomerization with other ERBB members, and activation of the PI3K/AKT/mTOR pathway. Cancers with NRG1 gene fusions have constitutive activation of NRG1 and HER2-HER3 pathways, and are sensitive to HER2/HER3 targeted therapies. 141 NRG1 gene fusions (with CD74, ATP1B1, and SDC4) are detected mostly in invasive mucinous lung adenocarcinomas and PDA (0.5%) and are enriched in KRAS WT tumors.142,143
Zenocutuzumab is a common light chain immunoglobulin G1 bispecific antibody with enhanced antibody-dependent cellular cytotoxicity (ADCC) that docks on HER2 and blocks the interaction between NRG1 and HER3. In a phase I/II trial for patients with refractory solid tumors harboring NRG1 fusions detected by RNA sequencing (77%), DNA sequencing (22%), or Nanostring (1%), zenocutuzumab conferred ORR of 34%, with median duration of response (DoR) of 9 months. 144 Among 19 PDA patients, ORR and DCR were 42% and 74%, respectively. Given meaningful efficacy with HER2/HER3-targeted therapies in cancers with NRG1 fusions, RNA sequencing should be included in molecular testing platforms.
BRAF
PDA has constitutive activation of the MAP kinase pathway due to gain-of-function mutations in KRAS, 145 but KRAS WT tumors can harbor RAS-independent RAF alterations: BRAF p.V600E in exon 15, BRAF p.N486_P490del in exon 11, and SND1-BRAF fusions, each present in 0.4–0.7% of pancreatic tumors. 15 In all, BRAF alterations are observed in 2% of PDA. 146
Evidence regarding benefit from BRAF-targeted therapy in PDA is expanding. In the phase II MyPathway basket study, four PDA patients with activating BRAF p.V600E mutations or other BRAF alterations were treated with the BRAF inhibitor vemurafenib, and one patient with a CUX1-BRAF fusion achieved a PR. 147 The Know Your Tumor registry included one patient with a BRAF p.V600E-mutated PDA who received matched therapy with BRAF/MEK inhibitors dabrafenib/trametinib and had a response for 11 months. 34 A retrospective case series of 81 PDA patients with RAF alterations including BRAF p.V600E (exon 15), BRAF ΔNVTAP (exon 11), and SND1-BRAF fusions showed variable benefit from BRAF/MEK-targeted therapies, with best responses (100%, 3/3 patients) observed for BRAF p.V600E-mutated PDA. 148 Atypical variants and multiple oncogenic drivers predicted lower/no response. The NCI-MATCH basket trial treated 35 solid tumors (3 PDA) harboring BRAF p.V600E mutations with dabrafenib/trametinib. One PDA patient had SD as best response. ORR was 35% among all patients, and median PFS and OS rates were 11.4 and 28.6 months, respectively, leading to the cancer agnostic FDA approval of this combination in pretreated cancers with BRAF p.V600E mutations. 149 A single-arm phase II trial is examining combined BRAF/MEK inhibition with encorafenib/binimetinib for pretreated advanced BRAF V600E-mutated PDA (NCT04390243).
RET
Rearranged during transfection (RET) proto-oncogene activates the downstream RAS/MAPK/ERK and PI3K/AKT pathways. 150 RET fusions can be detected with FISH, IHC, NGS, or RNA sequencing, and are most prevalent in papillary thyroid carcinomas (10–20%) and non-small-cell lung cancer (1–2%), 151 where RET inhibitors selpercatinib and pralsetinib gained FDA approval. RET fusions have been identified in 1% of PDA. 152 The phase I/II LIBRETTO-001 basket study with selpercatinib in advanced RET-altered (fusions or mutations) solid tumors included 11 PDA patients with ORR of 55%, 153 whereas in the phase I/II ARROW trial with pralsetinib for RET mutant/fusion positive tumors all four enrolled PDA patients responded (ORR: 100%), including one lasting 24 months+. 154 Selpercatinib was FDA approved for all solid tumors harboring RET fusions.
ALK, NTRK, and ROS-1
Anaplastic lymphoma kinase (ALK), neurotrophic tyrosine receptor kinase (NTRK), and c-ros oncogene 1 (ROS-1) fusions each have a prevalence of less than 1% and occur in KRAS WT PDA. 155 ALK fusions predict benefit from ALK inhibitors crizotinib, ceritinib, and alectinib. In a cohort of five patients with ALK-fusion positive advanced PDA (EML4-ALK and STRN-ALK), all of whom were KRAS WT and younger than 50 years, four patients received an ALK inhibitor, and three demonstrated SD, radiographic response, and/or normalization of serum CA 19-9. 155 Screening for ALK rearrangements should be considered in young patients with KRAS WT PDA.
TRK inhibitors, larotrectinib and entrectinib, have been FDA approved for tumors that harbor ROS1, NTRK1, NTRK2, and NTRK3 gene fusions. Among 159 patients with TRK fusion-positive cancers (2 PDA) treated with larotrectinib, ORR was 79% with a median DoR of 35.2 months and median PFS of 28.3 months. 156 In a pooled analysis of 121 patients with 14 tumor types (4 PDA) treated with entrectinib in the STARTRK-2, STARTRK-1, and ALKA-372-001 trials, the ORR was 61%, with a median DoR of 20 months and median PFS 13.8 months. 157 The NCCN guidelines recommend larotrectinib and entrectinib in PDA patients with NTRK gene fusions who have either failed prior therapies or in the first line setting if they have poor performance status and unfit to received conventional chemotherapy. 6 Like other targeted therapies, acquired resistance to these inhibitors develops. Second-generation pan-TRK inhibitors are being investigated, including selitrectinib (LOXO-195) and repotrectinib (TPX-0005, NCT03093116).
MET
Upregulation of the hepatocyte growth factor (HGF)/c-MET pathway occurs in more than 20% of PDA. HGF is produced by pancreatic stellate cells and its receptor, c-MET is expressed on epithelial PDA and endothelial cells. Elevated serum HGF levels have been reported to correlate with disease progression, 158 and high tumor c-MET expression is associated with poor survival. 159 Recently, a phase Ib study tested ficlatuzumab, a recombinant humanized anti-HGF antibody in combination with Gem-nabP as first-line treatment of 26 metastatic PDA patients, and showed acceptable tolerability (16% grade 3 hypoalbuminemia and 8% grade 3 edema), ORR of 29%, and median PFS and OS of 11 and 16.2 months. 160 Correlative biomarkers noted that responders had significantly higher baseline tumor pMET expression by IHC than non-responders (histoscore 80 versus 10, p = 0.047). While these data are encouraging, it is likely that combined ligand and receptor targeting would be needed for adequate targeting in future studies. 161
ARID1A
Mutations in epigenetic modifiers, such as the SWItch/sucrose non-fermentable component AT-rich interactive domain-containing protein 1A (ARID1A) promote the mesenchymal phenotype during pancreatic carcinogenesis. 162 ARID1A is a tumor suppressor harboring mutations in 2–8% of PDA. 163 ARID1A has also been implicated in double-stranded DNA repair via both homologous recombination and non-homologous end-joining, thought to confer platinum sensitivity when mutated. Loss of ARID1A leads to increased expression of the PI3K-interacting protein 1 gene (PIK3IP1), which downregulates PI3K-AKT signaling. 164 EZH2 inhibits PIK3IP1 gene transcription, and EZH2 blockade can upregulate PIK3IP1 upon ARID1A loss. 164 Other studies suggest that ARID1A-mutated cancers depend on HDAC activity and HDAC6 inhibition triggers cellular apoptosis. These mechanisms explain why epigenetic targeting of EZH2 methyltransferase with EZH2 inhibitors, and histone deacetylases with HDAC inhibitors for ARID1A-mutated cancers165,166 and are being investigated in PDA (NCT05053971). Lastly, there is a synthetically lethal interaction between ATR and ARID1A, and pre-clinical models have shown that ATR inhibition exploits a pre-existing DNA decatenation defect in ARID1A mutant tumor cells which causes premature mitotic progression. 167 M1774, an ATR inhibitor, is being tested in advanced solid tumors with loss of function in ARID1A (NCT04170153).
MDM2
The oncogene murine double minute 2 (MDM2) is overexpressed in up to 10% of PDA 168 and exerts its oncogenic activity via both p53-dependent and -independent pathways, promoting cancer cell growth and invasion and inducing resistance to chemotherapy. 169 MDM2 overexpression is a negative prognostic factor in PDA. 170 As a critical negative regulator of p53, MDM2 inhibition has growth inhibitory effects in p53 wild-type tumors. BI 907828, a highly potent MDM2-p53 antagonist is being studied in TP53 wild type, MDM2 amplified solid tumors, and preliminary data demonstrated clinical efficacy in sarcomas, pancreatic and biliary cancers (NCT03449381). 171
TP53 p.Y220C
Inactivating mutations in the tumor suppressor gene TP53 are common in pancreatic carcinogenesis and occur in 60% of PDA. Specific ‘hotspot mutations’, notably TP53 p.Y220C, affect the DNA binding domain and influence protein function. 172 Selective inhibitors designed against TP53 p.Y220C can stabilize p53 in the wild-type conformation, restoring transcription and tumor-suppressor function. 173 A phase I/II first-in-human study of PC14586 demonstrated an ORR of 24.2% in patients with advanced solid tumors, which included four patients with PDA (two SD, one PR). 174
FGFR
FGFR alterations occur in 8% of PDA, particularly in KRAS WT cancers. 15 In the phase I/II FIGHT-101 study evaluating the FGFR1–3 inhibitor pemigatinib in solid tumors harboring FGFR alterations, one of four PDA patients responded. 175 The RAGNAR phase II study recently reported on erdafitinib, a selective pan-FGFR TKI in 178 patients with advanced solid tumors with FGFR alterations. In all, 13 PDA patients had encouraging an ORR of 31%, a DCR of 85%, and a median DoR of 7.1 months. 176
Claudin 18.2
Claudin 18.2 is a tight junction protein expressed on normal gastric epithelial cells and overexpressed in several cancers including 16% of PDA. 177 While its role in cancer progression is poorly defined, because of its differential expression on cancer cells during carcinogenesis, Claudin 18.2 poses as a unique epitope to target. Zolbetuximab, a chimeric IgG1 monoclonal antibody which binds to Claudin 18.2 and mediates tumor cell death through ADCC, and complement-dependent cytotoxicity has shown promising activity in advanced gastroesophageal cancers in combination with chemotherapy. 178 A randomized phase II study is ongoing with Gem-nabP with and without zolbetuximab in patients with advanced PDA and high Claudin 18.2 expression (NCT03816163).
Claudin 18.2 also serves as a promising target for cellular immunotherapies, specifically chimeric antigen receptor (CAR) T cells. A phase I study evaluated a Claudin 18.2-directed CAR-T cell (CT041) in patients with pretreated gastroesophageal and other adenocarcinomas, including five PDA. An interim analysis showed an ORR of 49%, a DCR of 73%, and a 6-month OS of 80%. 179 Additional trials are being conducted among patients with gastroesophageal, PDA, and other gastrointestinal cancers (NCT04404595, NCT05539430). CD3 bispecific antibodies and ADCs against Claudin 18.2 have preliminary activity in gastrointestinal cancers, including PDA. 180 Multiple trials in advanced solid tumors, including PDA, are ongoing with Claudin 18.2-targeting bispecific antibodies (NCT04856150) and ADC (NCT04805307, NCT05009966, NCT05043987, NCT05161390).
Mesothelin
Mesothelin is highly expressed in many cancers, including PDA (⩾75–85%), with low levels expressed in healthy tissues. 181 Mesothelin activates the NF-κβ pathway, induces IL-6-mediated cancer cells proliferation, inhibits apoptosis, and stimulate invasion and migration via the p38 MAPK pathway. 182 These key roles highlight its importance in PDA progression, and the potential benefit of targeting this pathway.
A first-in-human clinical trial with anetumab ravtansine, an ADC of anti-mesothelin antibody linked to maytansinoid DM4, was conducted in patients with mesothelin expressing advanced solid tumors. 183 Among 148 patients enrolled, three of nine PDA (30%) patients had SD as best response. Durability of responses appeared to correlate with degree of mesothelin expression, with ⩾60% expression by IHC associated with the greatest benefit.
LMB-100, a recombinant immunotoxin (iTox) consisting of a mesothelin-binding fragment antigen-binding antibody region (Fab) for targeting and a modified Pseudomonas exotoxin A payload, in combination with nabP was tested in 20 refractory PDA patients. 184 While clinical activity was observed, the combination treatment was not tolerated due to capillary leak syndrome.
Cellular immunotherapy targeting mesothelin may be a promising approach, but has modest results to date. 185 A phase I study with mesothelin-specific CAR-T cells in six patients with chemotherapy-refractory metastatic PDA resulted in two patients achieving SD with PFS of 3.8 and 5.4 months, respectively. 186 Another phase I study of a single infusion of lentiviral-transduced mesothelin CAR-T cells in 15 subjects, including five with PDA, showed limited clinical activity with SD (11/15) as best response and only transient persistence of CAR T-mesothelin cells. 187 A limitation of CAR-T-cell therapy is its dependence on antigen expression on the cell surface. However, the majority (85%) of tumor-associated antigens and neoantigens are intracellular and are solely expressed in the context of an major histocompatibility molecule. 188 To sidestep this barrier, tumor antigen-specific T cells expressing a TCR can target intracellular proteins. 189 Preclinical studies in PDA noted benefit from TCR T cells targeting mesothelin, 190 and a phase I first-in-human study of autologous T cells expressing a high-affinity mesothelin-specific TCR is ongoing in refractory PDA (NCT04809766). Nevertheless, we recognize that T-cell exhaustion within the TME is likely to cause transient and suboptimal benefits and expect that further engineering of adoptive T cells to enable survival and effector function, while concurrent targeting of the immunosuppressive TME may lead to increased efficacy. 191
Conclusions
Large-scale genomic and transcriptomic analyses have provided unprecedented insight into the biology of PDA and promoted precision oncology by identifying novel targets and designing new drugs and combinations for targeted therapy (Figure 1). In all, 20–25% of PDA patients harbor targetable molecular alterations. Although PDA remains a devastating disease, by identifying subgroups of patients with actionable molecular alterations and applying biomarker-driven therapies, meaningful survival gains have been accomplished.