
Abstract
Recent explorations of the human gut microbiota suggest that perturbations of microbial communities may increase predisposition to different disease phenotypes. Dietary nutrients may be converted into metabolites by intestinal microbes that serve as biologically active molecules affecting regulatory functions in the host. Probiotics may restore the composition of the gut microbiome and introduce beneficial functions to gut microbial communities, resulting in amelioration or prevention of gut inflammation and other intestinal or systemic disease phenotypes. This review describes how diet and intestinal luminal conversion by gut microbes play a role in shaping the structure and function of intestinal microbial communities. Proposed mechanisms of probiosis include alterations of composition and function of the human gut microbiome, and corresponding effects on immunity and neurobiology.
Introduction
Gut microbiota and their effects on the human health
The concept of the human microbiome was first introduced to the scientific community by Joshua Lederberg, who defined it as ‘the ecological community of commensal, symbiotic, and pathogenic microorganisms that literally share our body space and have been all but ignored as determinants of health and disease’ [Lederberg and McCray, 2001]. Since the advent of the Human Microbiome Project (HMP), published studies describing the composition of the microbiota in normal and diseased human populations, along with novel methods in data mining and comparative analyses of the data, have been obtained. One intriguing discovery was demonstrated in a study that characterized fecal microbial communities of adult twin pairs concordant for leanness and obesity [Turnbaugh et al. 2009]. The results revealed that the human intestinal microbiome in each individual may share an identifiable core set of genes and pathways that are more highly conserved than microbial composition. Moreover, obesity was associated with changes in the microbiota at the phylum level and alteration in the representation of bacterial genes and metabolic pathways. Metagenomic analysis of data revealed a set of core microbial biomarkers of obesity involved in carbohydrate, lipid, and amino acid metabolism. Perturbations of core microbial functions, rather than core microbial communities, may be associated with alterations in physiological or disease states. Different approaches in comparative metagenomics include extensive queries against databases containing information on cellular functional networks. Such databases include the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) analyses. Results from these studies suggested that certain genetic elements from the intestinal microbiota may functionally complement the genes required for essential biological pathways in the human intestine that may have been missing or incompletely encoded in the human genome. Genes encoding functions involved in polysaccharide metabolism [Gill et al. 2006], methanogenic pathways for hydrogen gas removal, and enzymes for detoxification of xenobiotics [Kurokawa et al. 2007] have been identified as enriched functional genetic categories within intestinal microbial communities. Interestingly, a relatively comprehensive metagenomic gene catalog was published [Qin et al. 2010] and contained 3.3 million nonredundant microbial genes, assembled from paired-end reads of DNA isolated from the feces of 124 European individuals. The most recent data from the HMP report more than 5.2 million nonredundant genes [The Human Microbiome Project Consortium, 2012a], and aggregate estimates of the International Human Microbiome Consortium (IHMC) suggest that more than 8 million genes of the human microbiome have been discovered. This gene set is more than 300 times larger than the entire complement of genes in the human genome and contain a core of 24 ubiquitously present functional and metabolic modules among all samples, most of which consist of different enzyme families essential for microbial life [Abubucker et al. 2012]. Beyond metagenomes, scientists are exploring gene expression patterns in the human microbiome in order to understand functional metagenomics. A recent metatranscriptomic analysis of cDNA libraries prepared from fecal specimens of healthy volunteers demonstrated a common pattern of overrepresented genes, containing genetic elements involved in carbohydrate metabolism, energy production and synthesis of cellular components (Figure 1) [Gosalbes et al. 2011].
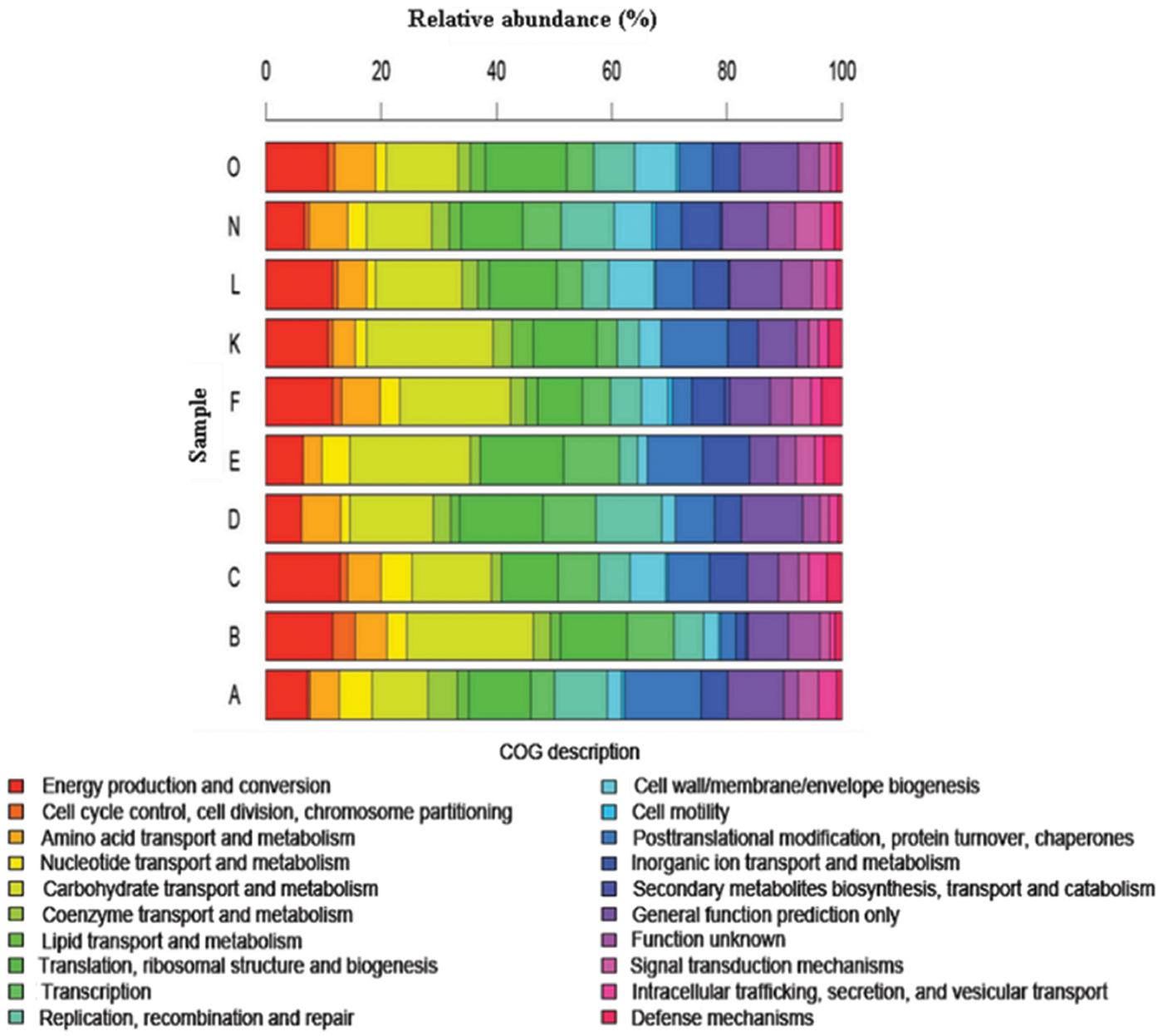
A recent metatranscriptomic analysis determined the distribution of functional roles of human fecal microbiota. This study demonstrated the distribution of Clusters of Orthologous Groups (COGs) categories across each of the 10 metatranscriptomes (A, B, C, D, E, F, K, L, N and O) that were sequenced. (Adapted from Gosalbes et al. [2011].)
Intestinal microbes can alter gene expression in the mammalian gut mucosa, ultimately affecting the function of the gastrointestinal tract. A study using germ-free and conventionally raised mice revealed that the gut microbiota modulated the expression of many genes in the human or mouse intestinal tract, including genes involved in immunity, nutrient absorption, energy metabolism and intestinal barrier function [Larsson et al. 2012]. Interestingly, most changes occurred in the mucosa of the small intestine. The presence of probiotics in the gastrointestinal tract can also affect patterns of gene expression, as demonstrated in a recent human study [Van Baarlen et al. 2010]. Healthy volunteers were subjected to treatment with probiotic bacteria (Lactobacillus acidophilus Lafti L10, L. casei CRL-431 and L. rhamnosus GG) and underwent esophagogastroduodenoscopy for duodenal specimen collection before and after a 6-week intervention period. Analysis of human gene transcriptional profiles in samples obtained from subjects treated with probiotics revealed changes in transcriptional networks involved in immunity and mucosal biology.
Diet and its effects on the gut microbiota
The possibility that diet may be able to influence the gut microbiota has been discussed in the scientific community since the 1960s. More recent studies have focused on using animal models and the analysis of intestinal microbiota and metagenomes to examine the association between diet and the composition and function of the gut microbiome. Human diets may have direct effects on the microbiome, which ultimately results in changes in the patterns of biochemical reactions in the intestinal lumen. In experiments using germ-free mice transplanted with human fecal microbiota, animals subjected to a high-fat, high-sugar Western diet demonstrated rapid changes in intestinal microbial community structure, with increased numbers of members of the phylum Firmicutes and decreased abundance of members of in the phylum Bacteroidetes. Metagenomic analysis revealed a concurrent overrepresentation of genes involved in carbohydrate transport. However, microbial communities returned to their original state within 1 week of switching back to a standard chow diet [Goodman et al. 2011]. This hypothesis led to the proposition of the ‘luminal conversion’ concept (Figure 2). Nutrients such as vitamins, amino acids or dietary fiber that are consumed by the host are assimilated and converted into other metabolites by intestinal microbes. Some products of these biochemical conversions, such as short-chain fatty acids (SCFAs), biogenic amines (such as histamine) or other amino-acid-derived metabolites such as serotonin or gamma-aminobutyric acid (GABA), may be biologically active in health and disease states. The production of these compounds may also be able to induce changes in microbial composition. Dietary nondigestible carbohydrates can be fermented in the intestinal lumen, resulting in production of SCFAs such as acetate, propionate and butyrate. Metabolically active SCFAs involved in many biological processes provide metabolic energy sources for human colonic epithelial cells. Moreover, fermentation of prebiotic carbohydrates such as inulin and fructo-oligosaccharides induces proliferation of beneficial microbes (mainly Bifidobacterium spp. and Lactobacillus spp.) in the gastrointestinal tract. Consumption of a fat-enriched diet has been suggested to affect the intestinal microbiota, as demonstrated in a recent clinical study whereby healthy volunteers were subjected to a high-fat, Western-style diet for 1 month. Plasma endotoxin levels were increased in individuals who were fed a high-fat diet compared with an isocaloric regular diet, which may be a result of perturbations in the gut microbiome [Pendyala et al. 2012]. However, it is still not known whether alterations of intestinal microbial communities represent causes or consequences of changes in human health status and different disease states.

Luminal conversion by intestinal microbes may play an important role in host–microbiota interactions. Orally consumed nutrients may be converted by intestinal microbes into bioactive compounds that could affect the health of the host and the intestinal microbiota. GABA, gamma-aminobutyric acid; SCFAs, short-chain fatty acids.
A recent study analyzed fecal metagenomes of individuals from different countries using multidimensional cluster analysis and principal components analysis (PCA) [Arumugam et al. 2011]. The authors were able to cluster the fecal metagenomes into three different enterotypes. These enterotypes were identified by relative amounts of any of three dominant genera: Bacteroides (enterotype 1), Prevotella (enterotype 2) and Ruminococcus (enterotype 3). Interestingly, these enterotypes appeared to be independent of nationality, sex, age or body mass index (BMI). However, results from a recent study [Wu et al. 2011] suggested that enterotypes may be strongly associated with long-term dietary composition. Bacteroides-enriched enterotype 1 was strongly associated with consumption of animal proteins and saturated fats, whereas Prevotella-enriched enterotype 2 was associated with a carbohydrate-based diet, consisting of simple sugars and fiber. Although it is not known whether enterotypes may be associated with predisposition to particular disease states, these findings suggested that long-term dietary patterns may affect enterotype status, the nutritional–microbiome connection and pathophysiology in disease-susceptible individuals. Alternatively, enterotypes may not be discrete and may represent enterogradients with differences in relative abundances of different taxa between individuals.
Definition of a ‘healthy’ gut microbiome
Data have recently emerged regarding the composition and function of the healthy gut microbiome. Stool specimens from 242 healthy, young adults were analyzed using 16S rRNA gene pyrosequencing and whole metagenomic sequencing to assess the composition and function of the microbiome, respectively [The Human Microbiome Project Consortium, 2012a, 2012b]. The biological diversity and richness of the distal intestine easily surpassed the relative richness of microbiomes, in terms of microbial taxa and genes, at other body sites such as human skin or the oral cavity. The predominant taxa varied in different body sites and, as expected, the phyla Bacteroidetes and Firmicutes represented the predominant phyla in the human intestine. The relative predominance of bacterial genera and species varied. For example, Bacteroides fragilis was present in a quantity of at least 0.1% of sequence reads in 16% of specimens from different individuals. Moreover, the prevalence of B. thetaiotaomicron was greater with quantities of at least 0.1% of sequence reads in 46% of individuals. Both Bacteroides species are known commensal intestinal taxa that have been cultured and studied in the laboratory for their immunomodulatory and metabolic features.
In contrast to microbial composition, whole genome metagenomics data demonstrated relatively even distribution and prevalence of metabolic pathways across body sites and individuals [The Human Microbiome Project Consortium, 2012a]. Predominant metabolic modules such as central carbohydrate metabolism represent major functional categories in the distal intestine as well as other body sites. However, 86% of identified families of genes from the gut metagenomes have not yet been functionally characterized or mapped to complete pathways [The Human Microbiome Project Consortium, 2012a].
Modification of the intestinal microbiota by the application of probiotics
Probiotics
According to the Food and Agricultural Organization of the United Nations and the World Health Organization, probiotics are defined as ‘living microorganisms, which when administered in adequate amounts confer health benefits on the host’ [Food and Agriculture Organization of the United Nations et al. 2006]. Nobel laureate Elie Metchnikoff introduced the concept of probiotics to the scientific community. He published a seminal report linking the longevity of Bulgarians with consumption of fermented milk products containing viable Lactobacilli [Metchnikoff and Mitchell, 1907]. This observation suggested that certain microbes, when ingested, could be beneficial for human health. Since then, probiotics had been widely marketed and consumed, mostly as dietary supplements or functional foods. Mechanisms of probiosis include manipulation of intestinal microbial communities, suppression of pathogens, immunomodulation, stimulation of epithelial cell proliferation and differentiation and fortification of the intestinal barrier (Figure 3) [Thomas and Versalovic, 2010].
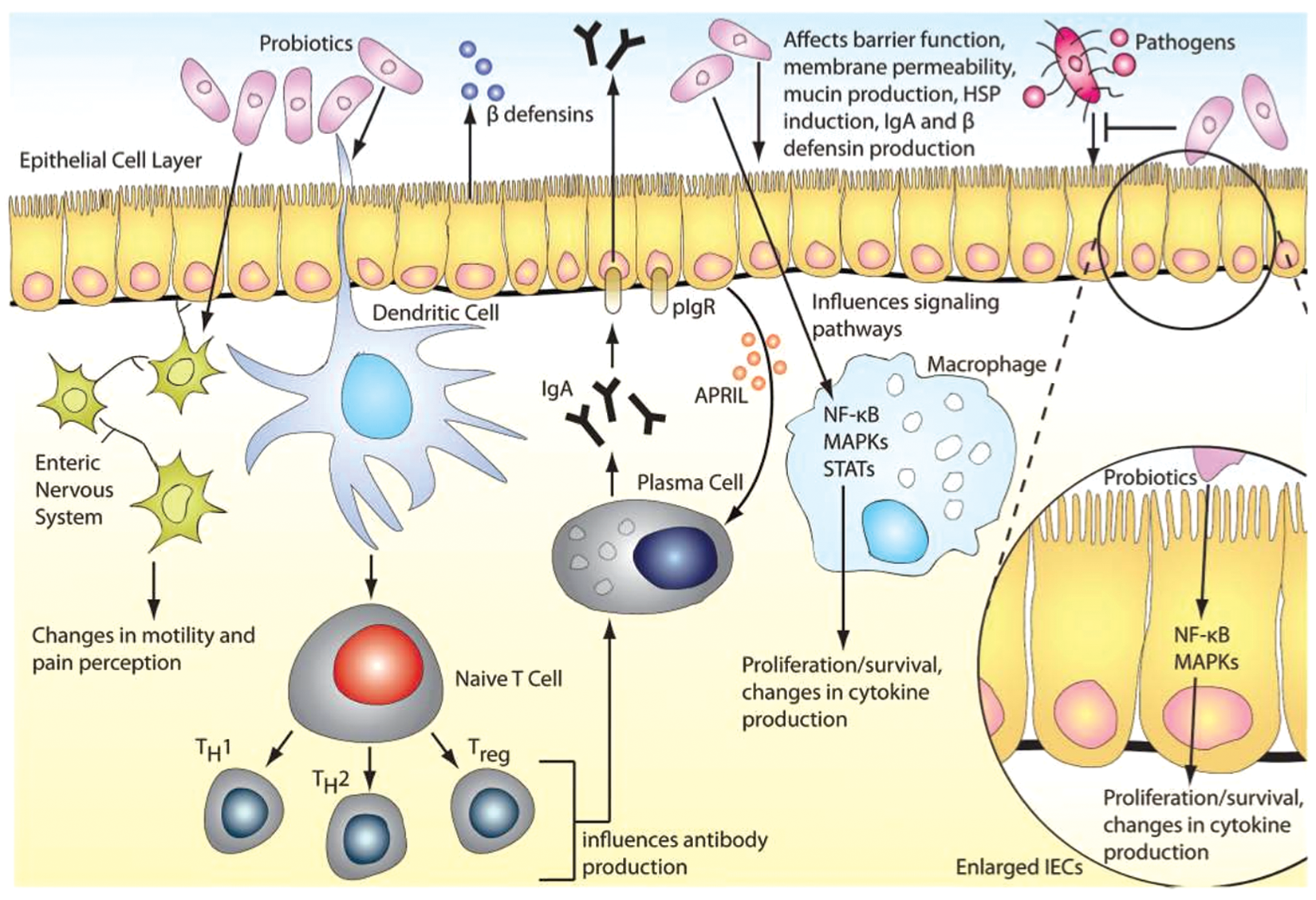
Probiotic mechanisms in the human gastrointestinal tract. Probiotics may manipulate intestinal microbial communities and suppress growth of pathogens by inducing the host’s production of β-defensin and IgA. Probiotics may be able to fortify the intestinal barrier by maintaining tight junctions and inducing mucin production. Probiotic-mediated immunomodulation may occur through mediation of cytokine secretion through signaling pathways such as NFκB and MAPKs, which can also affect proliferation and differentiation of immune cells (such as T cells) or epithelial cells. Gut motility and nociception may be modulated through regulation of pain receptor expression and secretion of neurotransmitters. APRIL, a proliferation-inducing ligand; hsp, heat shock protein; IEC, intestinal epithelial cell; Ig, immunoglobulin; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor-kappaB; pIgR, polymeric immunoglobulin receptor; STAT, signal transducer and activator of transcription; Treg, T regulatory cell. (Reproduced with permission from Thomas and Versalovic [2010].)
Dysbiosis and human diseases
The intestinal microbiome plays an important role in the function and integrity of the gastrointestinal tract, maintenance of immune homeostasis and host energy metabolism [Pflughoeft and Versalovic, 2012]. Perturbations in the composition of microbial communities, also known as dysbiosis, may result in disrupted interactions between microbes and its host. These changes in microbiome composition and function may contribute to disease susceptibility [Frank et al. 2011]. Several studies have demonstrated associations between intestinal dysbiosis and chronic low-grade inflammation [Cani and Delzenne, 2009] and metabolic disorders [Jumpertz et al. 2011], ultimately resulting in metabolic syndrome, obesity and diabetes [Claus et al. 2008; Larsen et al. 2010; Pflughoeft and Versalovic, 2012]. Alterations in the composition of the intestinal microbiome have been associated with infections in the gastrointestinal tract, inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS) [Pflughoeft and Versalovic, 2012; Saulnier et al. 2011]. Treatment modalities to manipulate and restore the balance in the richness and diversity of intestinal microbiome are being explored [Sonnenburg and Fischbach, 2012]. Probiotics may introduce beneficial functions into the gastrointestinal tract or enhance the functionality of existing microbial communities. Probiotics may also affect the composition and function of microbial communities by competition for nutrients, production of growth substrates or inhibitors and modulation of intestinal immunity [O’Toole and Cooney, 2008]. This concept is supported by results from randomized controlled clinical trials showing the benefits of probiotics during the treatment of gastrointestinal diseases (extensively reviewed by Preidis, Thomas and Versalovic [Preidis and Versalovic, 2009; Thomas and Versalovic, 2010]).
How probiotics alter the intestinal microbiota?
Proposed mechanisms of probiosis include effects on composition and function of the intestinal microbiome. Probiotics produce antimicrobial agents or metabolic compounds that suppress the growth of other microorganisms [Spinler et al. 2008; O’Shea et al. 2011], or compete for receptors and binding sites with other intestinal microbes on the intestinal mucosa [Collado et al. 2007]. Probiotic Lactobacillus strains enhance the integrity of the intestinal barrier, which may result in maintenance of immune tolerance, decreased translocation of bacteria across the intestinal mucosa, and disease phenotypes such as gastrointestinal infections, IBS and IBD [Lee and Bak, 2011]. Moreover, probiotics can modulate the intestinal immunity and alter the responsiveness of the intestinal epithelia and immune cells to microbes in the intestinal lumen [Thomas and Versalovic, 2010; Bron et al. 2011]. The effects of probiotics on the composition, diversity and function of the gut microbiota have been studied using different tools and techniques ranging from targeted, culture-dependent methods to metagenomic sequencing. However, not many studies have demonstrated associations of altered microbiota following treatment with probiotics. A clinical study demonstrated decreased pain and flatulence in patients with IBS that received a 4-week treatment with a rose-hip drink containing 5 × 107 colony-forming units (CFU)/ml of L. plantarum DSM 9843 per day [Nobaek et al. 2000]. This improvement in clinical symptoms was associated with the presence of L. plantarum in rectal biopsies of patients, along with reduced amounts of enterococci in fecal specimens. A more recent study focusing on patients with diarrhea-dominant IBS (IBS-D) yielded symptomatic relief in patients treated with a probiotic mixture of L. acidophilus, L. plantarum, L. rhamnosus, Bifidobacterium breve, B. lactis, B. longum and Streptococcus thermophilus. Interestingly, analyses of the fecal microbiota of these patients using denaturing gradient gel electrophoresis (DGGE) revealed that the similarity of the microbial composition was more similar in probiotics-treated patients than that of the placebo group. This observation suggested that microbial community composition was more stable during the period of probiotics treatment [Ki Cha et al. 2011].
Recent technological innovations in DNA sequencing and advancements in bioinformatics have provided scientists with tools to explore research questions related to the human microbiome and how treatment modalities affect changes in global composition and function of the microbial communities. A recent study [Cox et al. 2010] using a high-throughput, culture-independent method analyzed the fecal microbiota of 6-month-old infants treated with daily supplements of L. rhamnosus (LGG). Results showed an abundance of LGG and an increased index of evenness in the fecal microbiota of these infants, suggesting ecological stability. The ability of probiotics to induce changes in intestinal microbial communities was demonstrated by a recent study, which explored the effects of L. reuteri on microbial community composition in a neonatal mouse model using 16S rRNA metagenomic sequencing. Results from this study demonstrated a transient increase in community evenness and diversity of the distal intestinal microbiome in animals treated with L. reuteri compared with that of vehicle-treated animals [Preidis et al. 2012]. The diversity in microbial communities was shown to be associated with increased ecological stability [Eisenhauer et al. 2012]. The loss of species in a community, although not immediately visible, can result in diminished ecological resilience after a stress-related perturbation [Peterson et al. 1998]. Interestingly, reduced microbial diversity was associated with diseases such as Crohn’s disease [Manichanh et al. 2006] and eczema in early life [Forno et al. 2008]. Probiotics may induce changes in the intestinal microbiota and stabilize microbial communities. However, further studies in humans are needed to assess whether probiotics can make the same impact on the human intestinal microbiome and whether the changes are associated with clinical benefits in the host.
In addition to directly affecting the composition of the intestinal microbiota, probiotics may also modulate the global metabolic function of intestinal microbiomes. Fermented milk products containing several probiotics did not alter the composition of intestinal bacterial communities in gnotobiotic mice and monozygotic twins [McNulty et al. 2011]. However, fecal metatranscriptomic analysis of probiotics-treated animals demonstrated significant changes in expression of microbial enzymes, especially enzymes involved in carbohydrate metabolism. Moreover, mass spectrometric analysis of urinary metabolites revealed altered abundance of several carbohydrate metabolites. These observations suggested that probiotics may affect the global metabolic function of the intestinal microbiome.
Probiotics and intestinal immunomodulation
Dysbiosis may be associated with several human diseases, including IBD, infections and colorectal cancer [Karin et al. 2006; Artis, 2008]. Several probiotics modulate the intestinal immune system by production of secreted factors and metabolites that affect the growth and function of intestinal epithelial and immune cells (Figure 4) [Preidis and Versalovic, 2009].

Mechanisms of immunomodulation by beneficial microbes. Probiotics can modulate the immune system in the intestine through the luminal conversion process. The bacteria produce secreted soluble factors and metabolites, such as short-chain fatty acids (SCFAs) and vitamins using substrates from the diet. These bioactive compounds affect the function of intestinal epithelium and mucosal immune cells, resulting in production of cytokine and related factors such as a proliferation-inducing ligand (APRIL) and B-cell activating factor (BAFF). (Adapted from Preidis and Versalovic [2009].)
The Gram-positive bacterium L. reuteri is a heterofermentative symbiont indigenous to the gastrointestinal tract of many mammals including humans, pigs, mice and rats [Walter and Ley, 2011]. L. reuteri regulates the intestinal immune system by several different mechanisms [Lin et al. 2008]. Several in vitro studies have elucidated the molecular mechanisms behind its ability to modulate the immune system. Interestingly, these activities seem to be strain-dependent [Pena et al. 2004; Liu et al. 2010], and affect both innate and adaptive immunity.
Numerous studies have demonstrated the ability of L. reuteri to regulate cytokine production by human immune cells. Heat-killed L. reuteri 100-23 induced the production of anti-inflammatory cytokine interleukin (IL)-10 by bone-marrow-derived dendritic cells (BMDCs) [Livingston et al. 2009]. When these cells were treated with L. reuteri 100-23 and incubated with splenic T cells from ovalbumin T-cell receptor transgenic mice, IL-2 production was reduced and transforming growth factor-β (TGF-β) production increased, compared with that from nontreated cells. Moreover, Lactobacillus-free mice colonized with L. reuteri 100-23 contained more FoxP3-positive cells in their spleen and mesenteric lymph nodes than that of control mice. These results suggested that in addition to eliciting intestinal immune responses, L. reuteri 100-23 also regulated the development and recruitment of regulatory T cells to the gastrointestinal epithelium. L. reuteri may modulate gastrointestinal tract inflammation by regulating recruitment of different immune cells, as previously shown in gnotobiotic neonatal pig models of rotavirus infection. In piglets precolonized with human-derived L. reuteri ATCC 23272 and L. acidophilus NCFM, recruitment of monocytes and macrophages to the intestines and spleens was significantly reduced, compared with control animals. This result suggested that colonization by specific bacteria may ameliorate rotaviral-infection-induced pro-inflammatory immune cell recruitment to intestinal and systemic lymphoid tissues [Zhang et al. 2008].
Soluble factors from L. reuteri inhibited production of pro-inflammatory cytokines and signaling immune cells [Thomas et al. 2012]. Cell-free culture supernatants from murine-derived L. reuteri 6798 inhibited tumor necrosis factor (TNF) production by lipopolysaccharide (LPS)-activated [Pena et al. 2004] and Helicobacter hepaticus-treated [Pena et al. 2005] mouse macrophages. Moreover, conditioned media from human-derived L. reuteri ATCC PTA 6475 demonstrated the ability of strains to suppress TNF production by activated human monocytoid cells (THP-1) and primary monocyte-derived macrophages isolated from the peripheral blood of patients with Crohn’s disease. Transcriptional regulation of TNF expression by L. reuteri occurred via inhibition of c-Jun-dependent activator protein 1 (AP-1) signaling [Lin et al. 2008]. Moreover, L. reuteri was capable of forming biofilms, and TNF production was suppressed in human myeloid cells when THP-1 cells were treated with culture supernatants prepared from biofilms of L. reuteri PTA 6475 [Jones and Versalovic, 2009].
Comparative transcriptomics has identified several genes that may generate soluble, immunomodulatory factors [Saulnier et al. 2011]. The histidine decarboxylase gene (hdcA) is one such gene that encodes an enzyme that converts L-histidine into histamine. Interestingly, a recently published study from our lab has identified histamine as a potential immunomodulatory factor produced by L. reuteri ATCC PTA 6475. L. reuteri-derived histamine suppressed TNF production by THP-1 cells via stimulation of the H2 histamine receptor, ultimately resulting in suppression of TNF production by transcriptional regulation [Thomas et al. 2012]. Further characterization for other candidate genes and their potential roles in immunomodulation is ongoing.
The in vivo effects of L. reuteri in the human intestinal immune system was demonstrated in one study reporting the ability of L. reuteri ATCC 55730 to colonize the gastrointestinal tracts of healthy volunteers and patients with ileostomies [Valeur et al. 2004]. After supplementation with L. reuteri, duodenal B lymphocytes and CD4-positive T lymphocytes were significantly increased in the ileal epithelium, suggesting that probiotics stimulated the human adaptive immune system.
Probiotics and intestinal neuroimmunology
The human gastrointestinal tract contains a large and complex neural network called the enteric nervous system, whose main purposes are to regulate the physiological functions of the gut and modulate communication between the gut and the central nervous system, both in the ascending (gut-to-brain) and descending (brain-to-gut) directions [Sharma et al. 2009]. This communication system is called the gut–brain axis, which consists of intricate loops of neurological reflexes [Mayer, 2011]. The gut–brain axis moderates the coordination between the brain, the intestinal tract and endocrine and immune systems involved in maintaining gut function (Figure 5) [Bienenstock and Collins, 2010]. Disruptions or perturbations in the gut–brain axis have been associated with psychiatric symptoms such as anxiety and functional gastrointestinal disorders such as IBS [Neufeld and Foster, 2009].
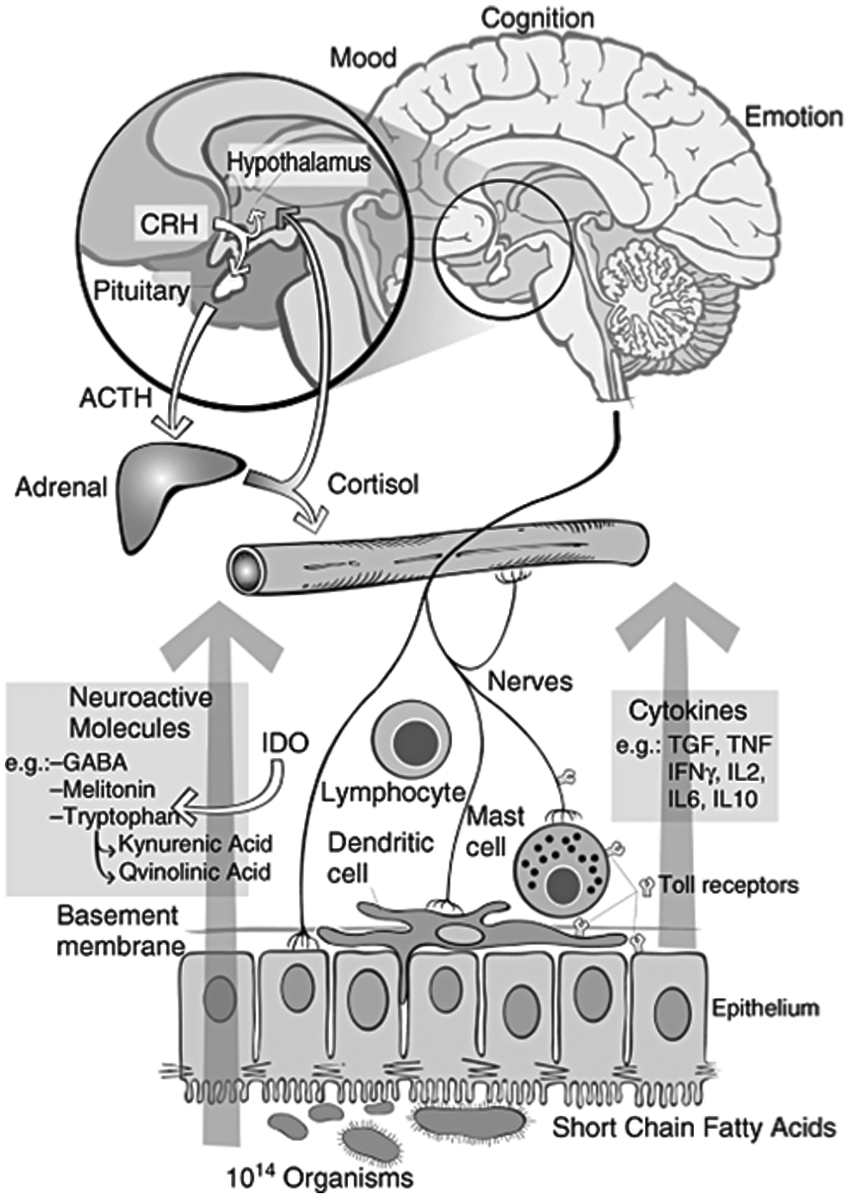
Proposed interactions between the gut microbiota, the intestinal tract, the central and peripheral nervous systems and the immune system. Intestinal microbes may interact with intestinal epithelial cells or immune cells directly or they can produce bioactive compounds and neurotransmitters to modulate immunity or the gut–brain axis. These intricate and complex interactions result in signaling to the central nervous system. CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone; IDO, indoleamine-pyrrole 2,3-dioxygenase. (Reproduced with permission from Bienenstock and Collins [2010].)
Investigations of the human microbiome and the development of next-generation sequencing technologies have yielded new opportunities for scientists to study the composition and function of the human microbiome and its association with neurological disorders. Hepatic encephalopathy, a condition commonly found in cirrhotic patients with liver failure and characterized by alterations in cognitive functions, was associated with changes in the gut microbiota and inflammation in the presence of intestinal barrier dysfunction [Bajaj et al. 2011]. Recent scientific studies shed light upon the intricate relationships between the intestinal microbiota and the gut–brain axis. Gut microbes may communicate with the gut–brain axis via production of neuroactive and neuroendocrine molecules such as serotonin, GABA, histamine, noradrenaline and adrenaline [Forsythe et al. 2009; Bienenstock et al. 2010]. A metabolomic study using germ-free mice demonstrated 2.8-fold higher amounts of serum serotonin in conventional mice compared with the level in germ-free mice, although direct evidence did not demonstrate production of serotonin by enteric bacteria [Wikoff et al. 2009]. However, other intestinal microbes such as Lactobacilli can convert glutamate into GABA [Higuchi et al. 1997; Li and Cao, 2010; Su et al. 2011], which functions as an inhibitory neurotransmitter in the central nervous system and may play a role in pain inhibition. Administration of L. rhamnosus JB-1 to mice resulted in altered patterns of GABA receptors in the brain, a reduction in stress-induced corticosterone and diminished anxiety- and depression-related behavior, all of which were absent in Lactobacillus-treated vagotomized animals [Bravo et al. 2011]. The inhibitory effect of gut bacteria on visceral pain originating in the gastrointestinal tract was demonstrated in a colorectal distension model in Sprague–Dawley rats. The treatment with L. rhamnosus ATCC 23272 for 9 consecutive days resulted in the inhibition of pain perception in animals when colonic distension was applied [Kamiya et al. 2006].
In addition to affecting the behavior and pain perception, interactions between intestinal microbes and the enteric nervous system was suggested to modulate immunologic responses in the intestine and extraintestinal sites. Gut microbes contribute to immune homeostasis and development (extensively reviewed by Jarchum and Pamer [Jarchum and Pamer, 2011]). Interestingly, intestinal microbes may contribute to the development of neuroimmunological disorders [Ochoa-Reparaz et al. 2011]. Investigations with a murine experimental autoimmune encephalomyelitis (EAE) model demonstrated the role of intestinal microbiota as a trigger for autoimmunity driven by myelin-specific CD4+ T cells [Berer et al. 2011]. Treatments with antibiotics [Yokote et al. 2008; Ochoa-Reparaz et al. 2009] or probiotics such as L. paracasei DSM 13434 and L. plantarum DSM 15312 [Lavasani et al. 2010] resulted in alleviation of EAE clinical symptoms and inflammation by suppression of IL-17 production and accumulation of regulatory T cells in secondary lymphoid organs. These observations suggested that probiotics could result in changes in the composition of the intestinal microbiome and favorable outcomes in patients suffering from autoimmune diseases.
Conclusion and future directions
Recent discoveries in the structure and function of the microbiome suggested that diet may have a direct impact on the intestinal microbiota and human or animal health status, and disruptions of microbe–man relationships may result in different disease states, including chronic inflammation, autoimmunity and neurological disorders.
Probiotics have been proposed as preventive and therapeutic measures, in order to restore the healthy composition and function of the gut microbiome. However, data from human microbiome studies may lead to identification of novel indigenous microbial species and tools to positively induce alterations in the gut microbial communities. Well-designed experiments in appropriate experimental models (in vitro or in vivo) may yield insights into the biology and potential manipulation of the microbiome in the human host. Metagenomic, metatranscriptomic and metabonomics can be deployed to globally examine interactions between probiotics, intestinal microbes and the mammalian gastrointestinal tract. New types of probiotics or medicinal compounds derived from the microbiome may be used as future strategies to promote health, prevent disease, and treat different disorders.
Footnotes
Funding
Our research cited in this article was supported in part by the National Institutes of Health to JV (R01 AT004326, R01 DK065075, UH3 DK083990). We also acknowledge NIH support (DK56338) for the Texas Medical Center Digestive Diseases Center.
Conflict of interest statement
J.Versalovic received unrestricted research support from Biogaia AB (Stockholm, Sweden).