
Abstract
Numerous clinical trials suggest that we have reached a limit in our ability to decrease the incidence of coronary heart disease (CHD) and cardiovascular disease (CVD) utilizing the traditional diagnostic evaluation, prevention and treatment strategies for the top five cardiovascular risk factors of hypertension, diabetes mellitus, dyslipidemia, obesity and smoking.
About 80% of heart disease (heart attacks, angina, coronary heart disease and congestive heart failure) can be prevented by optimal nutrition, optimal exercise, optimal weight and body composition, mild alcohol intake and avoiding smoking. Statistics show that approximately 50% of patients continue to have CHD or myocardial infarction (MI) despite presently defined ‘normal’ levels of the five risk factors listed above. This is often referred to as the ‘CHD gap’. Novel and more accurate definitions and evaluations of these top five risk factors are required, such as 24 h ambulatory blood pressure (ABM) results, advanced lipid profiles, redefined fasting and 2 h dysglycemia parameters, a focus on visceral obesity and body composition and the effects of adipokines on cardiovascular risk. There are numerous traumatic insults from the environment that damage the cardiovascular system but there are only three finite vascular endothelial responses, which are inflammation, oxidative stress and immune vascular dysfunction. In addition, the concept of translational cardiovascular medicine is mandatory in order to correlate the myriad of CHD risk factors to the presence or absence of functional or structural damage to the vascular system, preclinical and clinical CHD. This can be accomplished by utilizing advanced and updated CV risk scoring systems, new and redefined CV risk factors and biomarkers, micronutrient testing, cardiovascular genetics, nutrigenomics, metabolomics, genetic expression testing and noninvasive cardiovascular testing.
Introduction
Cardiovascular disease (CVD) remains the number one cause of morbidity and mortality in the United States. 1 The annual cost (direct and indirect) of treating CVD is approximately $320 billion USD. 1 Every one in three deaths is due to CVD with over 2200 US citizens dying from stroke or myocardial infraction (MI) daily.1–4 Clinical studies suggest that a limit has been reached in the ability to reduce CVD. 5 About 80% of coronary heart disease (CHD) can be prevented by optimal nutrition, optimal exercise, optimal weight and body fat, mild alcohol intake and avoiding smoking. 5 More than 400 CHD risk factors have been defined. 1 The three finite responses of the cardiovascular (CV) system to these insults include inflammation, oxidative stress and vascular immune dysfunction. Laboratory measurement of the finite responses allows the physician to ‘backtrack’ and determine the ‘why’ or the genesis of CVD, remove the insult(s) and initiate optimal prevention and treatment methodologies to resolve intermediate downstream abnormalities in order to meet defined clinical, noninvasive testing and laboratory goals. Prevention and treatment strategies must be directed to these finite responses while reducing the allostatic load of the 400 or more CHD risk factors and biomarkers and the intermediate downstream effects 1
The traditional diagnostic testing, evaluation, prevention and treatment strategies for the top five CV risk factors: hypertension, diabetes, dyslipidemia, obesity and smoking, have resulted in a ‘CHD gap’. 3 Approximately 50% of patients admitted to a hospital with acute coronary syndrome (ACS) or MI have ‘normal’ levels of the top five CHD risk factors.1,4 The ‘cholesterol-centric’ approach to prevent CHD is clearly important, but the other risk factors must be redefined, treated early and aggressively. Simultaneously, the clinician should measure and treat the three finite vascular responses. Advances in the direct assessment of the top five risk factors and the refinement of their definitions are often not utilized by physicians so that optimal prevention and treatment strategies for CHD are not clinically applied. 1 Physicians should also evaluate new CV risk scoring systems, novel and redefined CV risk biomarkers (factors), micronutrient testing, CV genetic nutrigenomics, metabolomics, genetic expression testing and noninvasive CV testing that allow nutritional medicine to become personalized and precise.
Revolutionizing the treatment of coronary heart disease
Addressing cell membrane physiology and identification of dysfunction represent the first step in the prevention and treatment of CHD. Cell membranes are the primary barrier between the external milieu (attacked by various hemodynamic or biochemical insults) and the internal cell organelles in the endothelium. The interaction of the various insults with the endothelial vascular receptors [pattern recognition receptors (PRR), nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), toll-like receptors (TLR) and caveolae] determine the signaling transduction into the cell. 1 The external insults stimulate these inflammatory vascular receptors directly or through epitopes. 1
Any cell membrane insult such as hypertension, alterations in hemodynamics, modified low-density lipoprotein (LDL) cholesterol, glucose, advanced glycosylation end products (AGE), microbes, internal tissue breakdown, toxins, heavy metals, homocysteine or the other CHD risk factors result in a reaction diffusion wave (tsunami effect) throughout the cell membrane that may disrupt cell receptors and the signaling mechanisms with subsequent membrane damage and dysfunction.6,7 Depending on the biological fatty acid makeup of the cell membrane with trans fatty acids (TFAs), saturated fatty acids (SFAs), monounsaturated fatty acids (MUFA), omega 3 fats (polyunsaturated fatty acids, PUFAs), the responses will vary. Fluidity within the cell membrane (MUFA, PUFA) dampens injury at the site of the insult, as well as throughout the rest of the cell membrane. However, stiff cell membranes (SFA, TFA) will exacerbate local and distant cell membrane damage. In addition, a heightened response (metabolic memory) from the inflammatory/immune cascade can create additional cell damage.6,7 The acute response to cell injury is defensive, short lived, regulated and appropriate, but with chronic insults of any type, the inflammatory, oxidative stress and immune responses become dysregulated and dysfunctional to the point that there is damage to the vascular system. In this regard, the blood vessel becomes an ‘innocent bystander’ to its own defense mechanisms that lead to functional then structural CV injury then preclinical CVD and clinical CVD. 1 The maladaptation of the renin–angiotensin–aldosterone system (RAAS), sympathetic nervous system (SNS) and the inflammatory/oxidative stress/immune pathways contribute to the vascular dysfunction. 1
The CHD risk factors have well-defined clinical goals that indicate the ‘normal’ level at which the risk for CHD is minimal. There exists a continuum of risk of CHD with all risk factors such that a ‘true’ normal level can be somewhat misleading.1,8–12 Normal blood pressure (BP) is defined as 120/80 mm Hg, prehypertension as 121/81–139/89 mm Hg and hypertension is defined as greater than 140/90 mmHg.11,13 Dyslipidemia is defined as a LDL-cholesterol (LDL-C) greater than 100 mg/dl, (depending on the clinical setting) and glucose intolerance at a fasting glucose over 99 mg/dl. 1 However, the continuum of risk starts at even lower levels for BP, LDL-C and glucose, as well as for most of the other CHD risk factors. 1 The risk for CHD actually starts with a fasting glucose of 75 mg/dl. For each 1 mg/dl increase in fasting blood sugar (FBS), the risk for CHD and MI increases by 1%.1,8–11 The risk for CHD starts at a BP level of 110/70 mm Hg. For every 20/10 mm Hg increase in BP the risk for CHD is doubled.1,11,11 The risk for LDL-C causing a reduction in endothelial nitric oxide (NO) is 60 mg/dl1,12
The concept of ‘translational vascular medicine’ correlates CHD risk factors with the actual presence of vascular disease. Do measured risk factors accurately translate into a vascular pathology that can be evaluated by noninvasive or invasive vascular testing? Conversely, does the absence of measured CHD risk factors accurately define the absence of vascular pathology or of vascular health? Evaluation of this correlation requires more sophisticated technology and advanced risk factor analysis combined with more comprehensive risk factor scoring systems such as those of the Consortium for Southeastern Hypertension Control (COSEHC)12,14 and Rasmussen 15 (Tables 1 and 2).
The Consortium for Southeastern Hypertension Control cardiovascular scoring system.

CV, cardiovascular; MI, myocardial infarction; LVH, left ventricular hypertrophy; FBS, fasting blood sugar; CHD, coronary heart disease; LDL, high-density lipoprotein; HDL, low-density lipoprotein.
Rasmussen cardiovascular scoring system.
(1) Disease score 0–2: no CV events in 6 years;
(2) Disease score 3–5: 5% CV events in 6 years;
(3) Disease score over 6: 15% CV events in 6 years;
(4) Superior to Framingham risk score;
(5) Variables measured: CAPWA, blood pressure at rest and exercise, LV mass by ECHO, microalbuminuria, BNP, retinal score, carotid IMT and ultrasound and EKG.

CV, cardiovascular; CAPWA, computerized arterial pulse wave analysis; LV, left ventricular; ECHO, echocardiogram; BNP, B-type brain natriuretic peptide; IMT, intimal medial thickness; EKG, electrocardiogram; BP, blood pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; A/V, arteriovenous; LVMI, left ventricular mass index; NT-pro BNP, N-terminal prohormone of B-type brain natriuretic peptide.
The endothelium, endothelial function, and endothelial dysfunction
The endothelium is a very thin lining of vascular cells forming an interface between the arterial lumen and the vascular smooth muscle (VSM).1,3,16 Endothelial dysfunction occurs when NO bioavailability is reduced, which leads to inflammation, oxidative stress, immune dysfunction, abnormal growth, vasoconstriction, increased permeability, thrombosis and CHD.1,3,16,17
LDL-C has a primary role in the development of atherosclerotic plaque formation starting at birth (Figure 1). 18 LDL-C migrates from the blood into the subendothelial layer, attaches to proteoglycans and becomes modified, antigenic and toxic damage-associated molecular patterns primarily via oxidation of LDL (oxLDL) and glycation of LDL (glyLDL) occur. 18 The modified LDLs produce numerous cytokines and chemokines that attract monocytes into the subendothelial layer which transform into macrophages. The modified LDLs are taken by scavenger receptors (SRAs) on the macrophage cell membranes, which transform into foam cells then fatty streaks and eventually form a coronary artery plaque (stable plaque or a rupture-prone plaque) that can result in an MI. Approximately 45 different steps exist in this process of dyslipidemia-induced vascular disease that can be interrupted with nutrition, nutritional supplements and drugs. 18 Endothelial dysfunction, arterial pathology, cardiac dysfunction and CHD represent a delicate balance of vascular injury (angiotensin II and endothelin) and nitric oxide and vascular repair from endothelial progenitor cells, produced by the bone marrow.1,3,6

The pathogenesis of atherosclerotic plaque formation.
The pathophysiology of vascular disease
The primary causes of CHD are the three finite responses that are generated from the large allostatic environmental insults coupled with CV genetics, nutrigenomics, metabolomics, proteomics and gene expression patterns. The three finite vascular responses are:
Figure 2 illustrates the interconnection of the external insults and the vascular receptors (PRR, NLR, TLR, caveolae) on the endothelium. These insults are divided into two major categories: biomechanical (BP, pulse pressure, shear stress and oscillatory pressure) within the arterial system and external biochemical factors that include dietary factors, various bio-humoral and metabolic factors, microbes, sterile and nonsterile antigens, and environmental toxins. 19
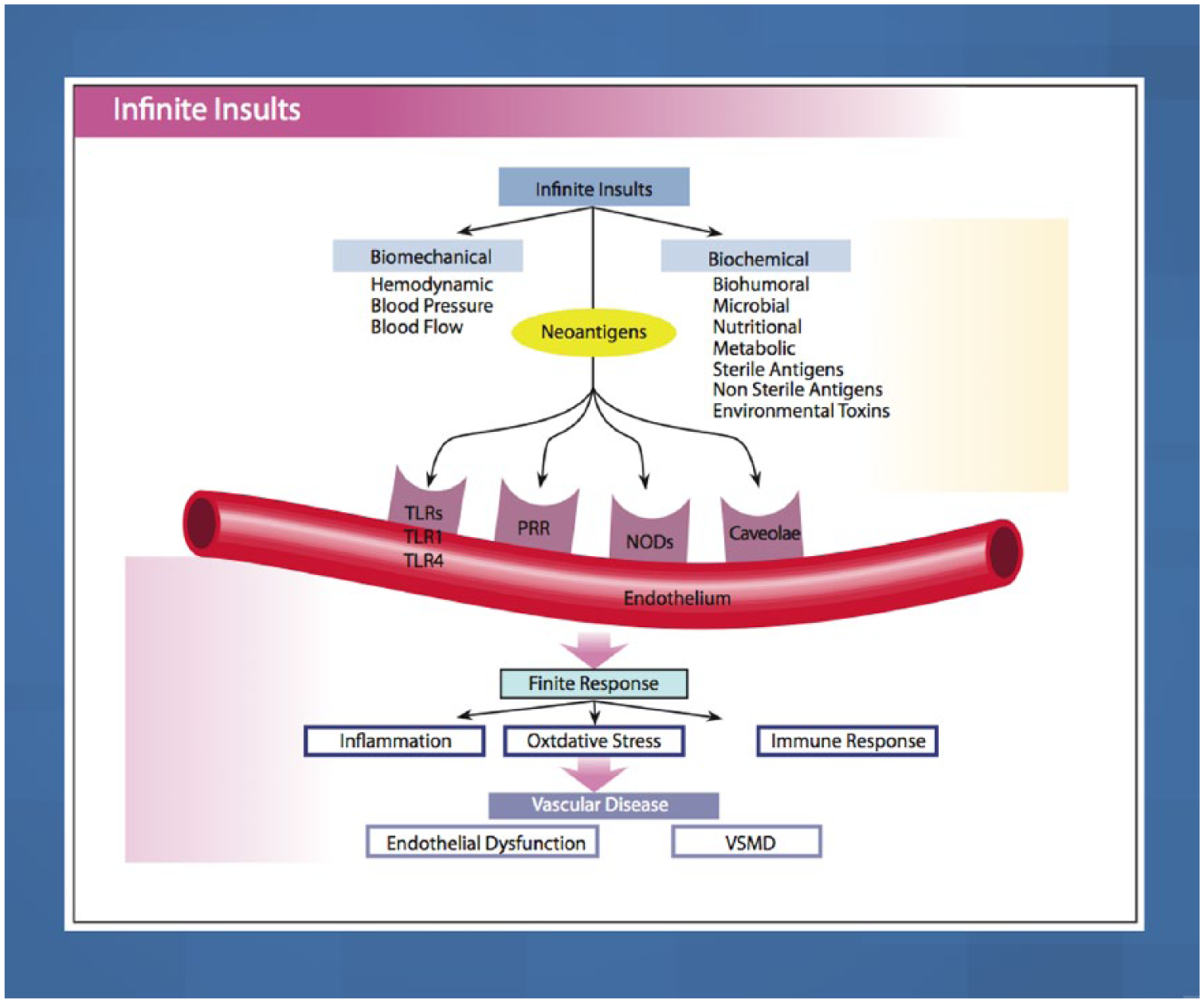
Biochemical and biomechanical insults interact with vascular receptors, PRRs, NLRs, TLRs and caveolae, to induce the three finite responses of vascular inflammation, oxidative stress and vascular immune dysfunction which lead to endothelial dysfunction, and VSM and cardiac dysfunction, coronary heart disease, myocardial infarction and congestive heart failure.
Interrupting the finite pathways
The reduction of the allostatic load, interrupting the insult–vascular receptor interaction to the PPR, NLR, TLR and caveolae, and disruption of the downstream mediators are paramount to a successful prevention and treatment regimen for CVD. Numerous scientifically validated nutritional or dietary components and nutraceutical supplements have great promise in this regard. 20 These will be discussed in detail in the treatment section.
Preventing and treating CHD and establishing CV ecology and balance involve utilizing a more complex and logical approach, such as dynamic systems biology, functional and metabolic medicine. As one might expect with a complex network of physiological interactions underlying vascular responsiveness and development of CHD, a single genetic cause has not been identified. Instead, as many as 30 separate loci are associated with MI and CHD. The majority of these involve inflammatory pathways but only a minority of those 30 loci relate the top five CV risk factors. 2
Atherosclerosis and endothelial dysfunction
Atherosclerosis and endothelial dysfunction are postprandial diseases. 21 The consumption of sodium chloride (NaCl), refined carbohydrates (CHO), sugars, starches and some, but not all, SFAs and TFAs will promote glucotoxicity, triglyceride toxicity, vascular metabolic endotoxemia, inflammation, oxidative stress, and vascular immune dysfunction that may persist long after the initial insult. This may also result in an exaggerated response (metabolic memory) with repeated or chronic dietary insults.6,21 Fortunately, studies have shown that eating a diet rich in low-glycemic foods, reduced refined sugars and starches, low in NaCl and high in potassium and magnesium, high in MUFAs, PUFAs, polyphenols, and antioxidants can help to prevent the postprandial endothelial dysfunction and endotoxemia. 18 Early evidence of CHD in the form of fatty streaks in the aorta and coronary arteries has been documented in children in the first and second decades of life and in postmortem exams of teenagers and war victims (Figure 3). 1 The CV disease is subclinical for decades prior to any CV events.1,3,16 Endothelial dysfunction is the earliest functional abnormality, followed later by changes in arterial compliance of small resistance arteries and then larger conduit arteries, with loss of elasticity leading to hypertension, VSM hypertrophy, diastolic dysfunction (DD), left ventricular hypertrophy (LVH), congestive heart failure (CHF) and CHD. 17

Atherosclerosis progression.
Figure 4 shows the progression from subintimal coronary atherosclerosis to obstructive CHD. The coronary artery on the left is normal, the one in the center demonstrates mild subintimal disease with increased intimal medial thickness (IMT) but a normal and unchanged arterial lumen. This extraluminal disease and inflammation would be captured using computed tomography angiogram (CTA) or intravascular ultrasound (IVUS) but missed by conventional coronary arteriogram (Figure 5). The image on the right in Figure 4 illustrates extensive extraluminal and intraluminal obstructive CHD. Most MIs occur with mild stenosis of the coronary arteries.
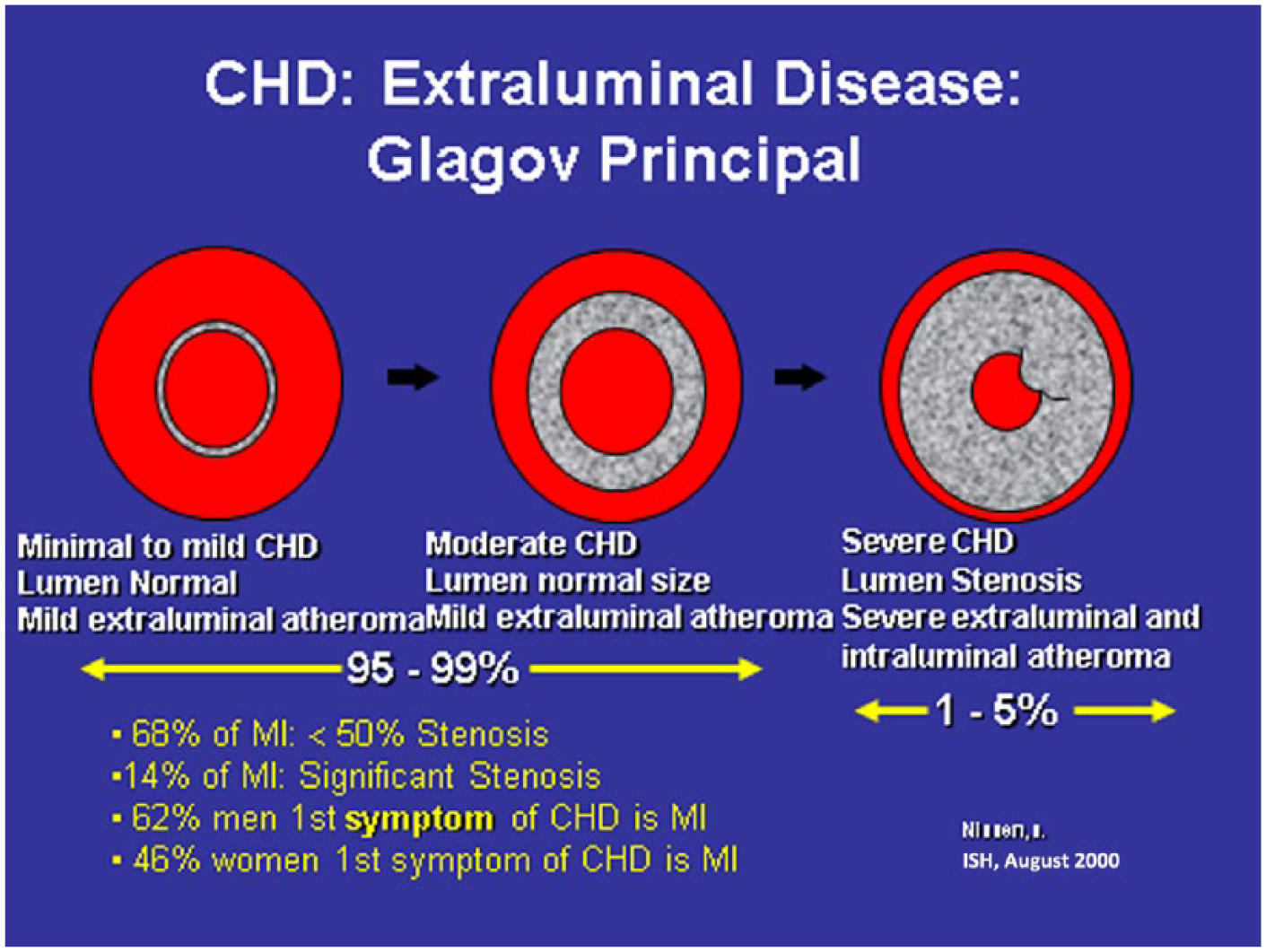
Progression of subintimal coronary atherosclerosis to obstructive coronary heart disease.

Coronary heart disease that is not detectable by angiogram (left) is evident using intravascular ultrasound (right).
Some of the top coronary heart disease risk factors and the coronary heart disease gap
The ‘CHD gap’ is related to incorrect definitions, assessment and treatment of the top five risk factors, the lack of assessment and treatment of the other 395 risk factors, not performing the various noninvasive CV tests, genomic CV individuality and possibly other unknown factors. 1
Hypertension
Only a 24 h ABM accurately measures and predicts CHD.1,16 Measurements of nocturnal BP and dipping status (normal is a 10% mean reduction from the daytime BP mean to the night), nondipping status, BP surges, BP load (normal is below 140/90 mm Hg in 15% of the total BP measurements) and BP variability are superior to office BP readings as a predictor of CHD risk.1,16 Excessive dipping is associated with an increased risk of ischemic stroke and reverse dipping is associated with an increased risk of intracerebral hemorrhage (ICH).1,16 Nocturnal blood pressure is more clinically important than daytime blood pressure (a 27/15 mmHg difference is optimal).1,16 Morning blood pressure surges (level and rapidity) increase the risk of ischemic stroke, MI, and LVH.1,16 Hypertension is a marker for vascular endothelial dysfunction with reduced NO bioavailability, but the vascular disease is enhanced in a bidirectional manner with hypertension which leads to greater vascular damage. 17 The items that should always be considered when evaluating blood pressure include: 16
A normal blood pressure is 120/80 mmHg, but there is a continuum of risk for CHD starting at 110/70 mmHg;
Each increase of 20/10 mmHg doubles CHD risk;
Before age 50, the diastolic blood pressure is a better predictor of CHD risk;
After age 50, the systolic blood pressure (SBP) is a better predictor of CHD risk;
24 h ABM is more accurate than office blood pressure measurements and should be the standard of care for defining blood pressure and CHD risk;
Mercury sphygmomanometers are preferred. Electronic arm units are accurate if done correctly and validated. The wrist or finger monitors are not as accurate and should not be used as the basis for a hypertension diagnosis.
Dyslipidemia
Dyslipidemia should be evaluated using advanced lipid profiles to determine treatment and predict individual CHD risk more accurately.18,22,23 An advanced lipid profile will measure:
Total LDL-C;
LDL-P (LDL particle number) which drives CHD risk;
LDL size (the dense type B LDL is more atherogenic versus large type A LDL);
Modified LDL (oxidized, glycated, glyco-oxidized and acetylated);
Apolipoprotein (APO) B and APO A;
Lipoprotein a [Lp(a)];
Total high-density lipoprotein (HDL);
HDL particle number (HDL-P);
HDL size and HDL mapping (large 2b versus small type 3) or five types of HDL;
Dysfunctional HDL;
Reverse cholesterol transport and cholesterol efflux capacity (CEC);
Myeloperoxidase (MPO);
APO-CIII;
VLDL and triglyceride (TG) total;
Large very low-density lipoprotein (VLDL);
VLDL-P particle number;
Remnant particles.
The primary CHD risk related to LDL-C is LDL-P and APO B particles.12,18,22,23 Dense LDL is also predictive but only if LDL-P is elevated above the normal level.12,18,22,23 oxLDL is also associated with increased CHD. HDL-P is more protective for CHD with larger HDL type 2b being a second important protective mechanism.12,18,22,23 Greater number and size of HDLs are more efficient at reverse cholesterol transport, as well as having numerous other protective effects such as reducing inflammation, oxidative stress and immune dysfunction. It is also important to analyze dysfunctional HDL with MPO.12,18,22,23 Patients who have an HDL of 85 mg/dl or more, often have dysfunctional HDL that may not be protective.22,23 The VLDL, especially large VLDL, triglycerides and remnant particles are very atherogenic and thrombogenic. 18
Dysglycemia, insulin resistance and diabetes mellitus
An FBS of over 75 mg/dl increases CHD risk by 1% per increase of 1 mg/dl of FBS and induces endothelial dysfunction.1,8–10 A 2 h glucose tolerance test (GTT) over 110 mg/dl increases CHD risk by 2% per 1 mg/dl over the 110 mg/dl level.1,8–10 The current definition of an abnormal 2 h GTT is >140 mg/dl. Hyperinsulinemia is also an independent risk factor for CHD.1,8–10 Calculating a homeostasis model assessment (HOMA) score will provide additional insight into the clinical presence of insulin resistance. Multiplying the FBS by the insulin level and dividing by 400 will give an excellent estimate of HOMA. A normal HOMA is <1.0, mild insulin resistance is between 1.0 and 2.0 and over 2.0 is severe insulin resistance. Insulin resistance creates inflammation, reduces NO levels, causes endothelial dysfunction and vascular disease through the mitogen-activated protein kinase (MAPK) pathway, which is a hypertensive, inflammatory and proatherogenic pathway. 1 On the other hand, the phosphatidylinositol 3-kinase (PI3K) pathway is anti-inflammatory, antihypertensive and antiatherogenic. 1
Noninvasive testing
Fortunately, there are many noninvasive tests available to determine CV pathology prior to clinical CHD 1 (Table 3). One of the best validated early detection tests for functional abnormalities of the endothelium is the EndoPAT which determines endothelial function and dysfunction.24–26 The EndoPAT measures postocclusion brachial artery hyperemia, which is an excellent indirect measure of NO bioavailability and endothelial dysfunction in the coronary arteries. The EndoPAT predicts accurately the future risk for hypertension, CHD, unstable angina, CVD, CHF, MI, cardiac death, hospitalization, coronary artery bypass graft, stent restenosis, the presence of plaque in the coronary arteries that are rupture prone, pulmonary artery disease (PAD) and cerebrovascular accident (CVA) beyond the Framingham risk scoring (FRS).25–27 In a study of 528 patients with high risk for CV events over 5 years, the EndoPAT reactive hyperemia index (RHI) was measured before and after coronary angiogram. 28 The RHI, brain natriuretic peptide (BNP) and CV score by SYNTAX were independent risk predictors for all future CV events such as MI, CV death, unstable angina, ischemic CVA, coronary artery bypass graft (CABG), CHF and PAD.
Noninvasive cardiovascular testing.
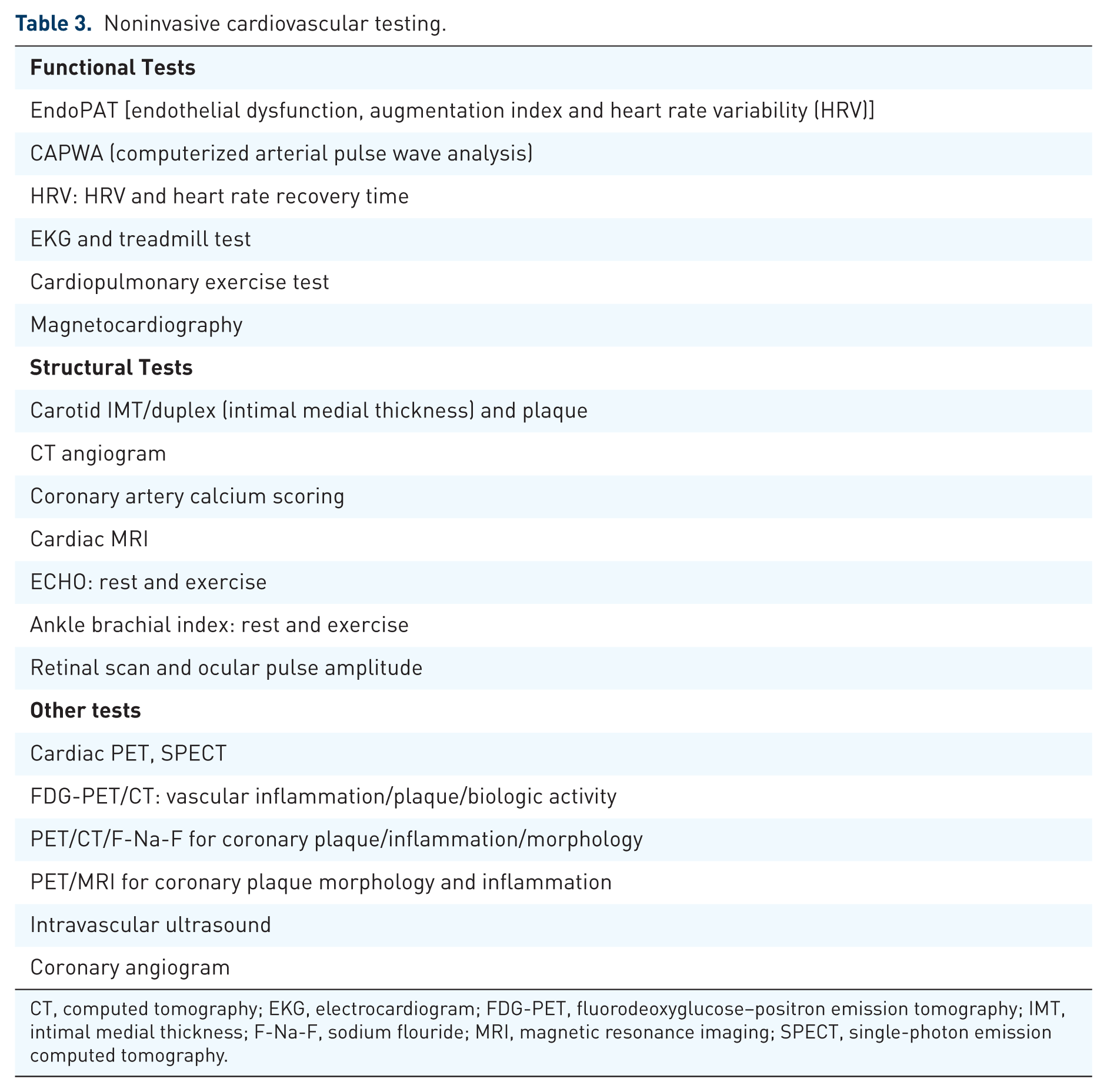
CT, computed tomography; EKG, electrocardiogram; FDG-PET, fluorodeoxyglucose–positron emission tomography; IMT, intimal medial thickness; F-Na-F, sodium flouride; MRI, magnetic resonance imaging; SPECT, single-photon emission computed tomography.
When RHI was added to FRS, BNP and SYNTAX, the net reclassification index was significantly improved by 27.5% with a significant increase in C-statistic from 0.728 to 0.766. A normal RHI is over 1.67. 28
The computerized arterial pulse wave analysis (CAPWA) (CV profiler) also predicts future CHD by measuring large and small arterial compliance.29–32 The C2 compliance identifies the presence of endothelial dysfunction in the microvascular circulation, the very small arterioles and medium-sized arteries (range 4–9 microns). The C1 compliance is a measure of the elastic behavior of the aorta and larger arteries (range 8–17 microns). Lower numbers of C1 and C 2 compliance indicate diseased arteries, arterial stiffness, decreased arterial compliance of the vascular wall and endothelial dysfunction. These are all age and gender adjusted. The CAPWA improves risk stratification beyond usual risk factors, including microalbuminuria (MAU), ECHO and carotid IMT. A low C2 and increased pulse wave velocity predict CHD.
The carotid IMT predicts future risk of CHD and CVA.33–35 Normal values without any plaque present must be adjusted for age and sex. A carotid IMT of <0.6 mm is normal to low risk, 0.6–0.7 mm is moderate risk, and 0.7–0.95 mm is high risk for future CVD. The normal IMT accretion rate (CIMTAR) is <0.016 mm/year. Carotid IMT indicates preclinical atherosclerosis and intimal-medial hypertrophy secondary to vascular smooth muscle and fibrous cell hypertrophy and hyperplasia. Carotid IMT correlates well with CHD risk factors and future CV events such as CHD, MI, transischemic attack (TIA) and stroke. The risk for MI is 1.26 [95% confidence interval (CI) 1.21–1.30] per 1.0 standard deviation (SD) of common carotid artery IMT difference and 1.15 (95% CI 1.12–1.17) per 0.10 mm of common carotid artery IMT difference over 5 years. 35 The risk for stroke is 1.32 (95% CI 1.27–1.38) per 1.0 SD common of carotid artery IMT difference and 1.18 (95% CI 1.16–1.21) per 0.10 mm common of carotid artery IMT difference over 5 years. 35
Fundus examination of retinal arterioles with SLDF (scanning laser doppler flowmetry) correlates highly with micromyographic biopsies of the medial lumen ratio (MLR) in subcutaneous small arteries. Retinal pathology indicates microvascular disease even after adjustment of renal dysfunction and traditional CVD risk factors. Retinal microvascular endothelial dysfunction assessed with flicker light of retinal veins and arteries is a nitric-oxide-dependent phenomenon and predicts hypertension, as well as CVD and CHD.36–39
Coronary artery calcification (CAC) was associated more strongly than carotid IMT with the risk of incident CHD in 6698 subjects over 5.3 years. The CAC predicted a CHD risk increase of 2.1-fold per 1.0 SD, whereas the carotid IMT predicted a CHD risk increase of 1.3-fold per 1.0 SD. 40 CAC progression over 15% annually provides increased CHD risk analysis with a 17-fold increase in CHD. A baseline CAC score predicts CHD risk beyond traditional risk factors and a CAC score of over 300 has a hazard ratio of CHD of 10. A positive CAC increases risk of major cardiac events by 6–35-fold. CAC correlates with traditional risk factors but also with increased oxidative stress, autoantibodies to oxLDL, APO B-immune complexes and glycemic load and index.41–44
The risk of major CV events or death increases in a graded manner with the degree of coronary atherosclerosis as defined by CTA, even in the absence of high-grade coronary artery stenosis. Coronary CTA detects approximately twice as many coronary segments with plaque compared with coronary angiograms. This results in 52% of patients being assigned to a greater risk category.45–47
The multifunction cardiogram (MCG) is a computational electrophysiologic system to detect abnormal stress and strain between the myocardium (visco-elastic solid) and intracardiac blood flow (non-Newtonian fluid at low and intermediate shearing states) from a two-lead (II and V5) resting electrocardiogram (EKG). The MCG detects myocardial ischemia in an 82 s analysis.48–51 This maps the heart’s electrical activity to predict early CHD, ACS, MI and arrhythmias. The sensitivity is 88% and specificity is 88% (range 80–100%) for the early diagnosis of CHD, depending on degree of stenosis in some studies,50,51 but less specific and sensitive in another trial. 49
Other noninvasive CV tests include the cardiac exercise ECHO, CEPT (cardiopulmonary exercise testing), exercise treadmill, various cardiac nuclear medicine scans, magnetic resonance imaging (MRI)/magnetic resonance angiography (MRA) for coronary plaque and obstruction, the fluorodeoxyglucose–positron emission tomography (FDG-PET)/computed tomography (CT) for arterial inflammation that defines biologic activity, as FDG accumulates in activated immune cells such as macrophages and T cells due to increased glycolysis. It predicts CAC/CHD events. The PET/CT/sodium flouride (F-Na-F) predicts coronary thrombosis, plaque and inflammation and the PET/magnetic resonance (MR) predicts coronary artery plaque and inflammation.52,53
The micronutrient test for functional deficiencies of nutrients (SpectraCell, Houston, TX, US) is valuable for assessing nutritional status and provide a more scientific rationale for nutrition and nutritional supplement treatment for CHD and hypertension. 54 This is a lymphocyte assay that measures the status of 28 micronutrients for the previous 6 months. A recent study found that approximately 62% of hypertensive subjects could taper or discontinue pharmacologic therapy utilizing an aggressive micronutrient replacement program in conjunction with other lifestyle changes. 54
Treatment
Nutrition
Mediterranean diet: traditional Mediterranean diet
In the 4.8-year primary prevention trial (PREDIMED), the rate of major CV events from MI, CVA or total CV deaths was reduced by 30% with nuts and 30% with extra virgin olive oil (EVOO). The reduction in CVA was 39% (p < 0.003) with a 33% reduction from EVOO and a 46% reduction from nuts. The reduction in MI was 23% (p = 0.25) with a 20% reduction from EVOO and a 26% reduction from nuts. Total CV deaths were reduced by 17% (p = 0.8).55–58 New-onset type 2 diabetes mellitus (T2DM) was decreased by 40% with EVOO and 18% with mixed nuts. 58 This reduction was associated with decreases in hsCRP and interleukin-6 (IL-6).
The high content of nitrate (NO3), at an average of 400 mg per day, is converted to nitrite (NO2), which eventually forms NO. Also, the increased amount of omega 3 fatty acids (FAs), good omega 6 FAs and polyphenols (such as quercetin, resveratrol and catechins, in grapes and wine) provide many of the beneficial outcomes in CHD. 57 Secondary prevention post MI in the Lyon Heart Study demonstrated significant reductions in all events including cardiac death, nonfatal MI, unstable angina, CVA, CHF and hospitalization at 4 years 59 using the Mediterranean-style diet supplemented with alpha-linolenic acid compared with a prudent western diet. Compared with the control, the Mediterranean-style diet demonstrated a 76% lower risk of cardiac death and nonfatal MI during the study period. 59 Olive oil was associated with a decreased risk of overall mortality and an important reduction in CVD mortality in a large Mediterranean cohort of 40,622 participants. For each increase in olive oil by 10 grams per day there was a 13% decrease in CV mortality. In the highest quartile of olive oil intake, there was a 44% decrease in CV mortality. 60 One of the mechanisms by which the traditional Mediterranean diet (TMD), particularly if supplemented with virgin olive oil at 50 grams per day, can exert CV health benefits is through changes in the transcriptomic response of genes related to CV risk that include genes for atherosclerosis, inflammation, oxidative stress, vascular immune dysfunction, T2DM and hypertension. This includes genes for ADR-B2 (adrenergic beta 2 receptor), IL7R (interleukin 7 receptor), IFN gamma (interferon), MCP1 (monocyte chemotactic protein), TNFα (tumor necrosis factor alpha), IL-6 and hsCRP.56,61–63 In summary, the TMD has been shown to have the following effects:55,56,61–63
Lowers BP;
Improves serum lipids: lowers total cholesterol (TC), LDL, TG, increases HDL, lowers oxLDL and Lp(a), improves LDL size and lowers LDL-P to a less atherogenic profile;
Improves T2DM and dysglycemia;
Improves oxidative defense and reduces oxidative stress: F-2 isoprostanes and 8 hydroxy guanosine;
Reduces inflammation: lowers hsCRP, IL-6, soluble vascular adhesion molecule, soluble intercellular adhesion molecule;
Reduces thrombosis and factor VII after meals;
Improves BNP;
Increases nitrates/nitrites;
Improves membrane fluidity;
Reduces MI, CHD and CVA;
Reduces homocysteine.
Dietary approaches to stop hypertension diets (DASH) (DASH 1 and 2)
The dietary approaches to stop hypertension (DASH) diets reduce BP and CHD. Both DASH 1 and DASH 2 emphasize fruits, vegetables, whole grains, beans, fiber, low-fat dairy products, poultry, fish, seeds and nuts, but limit red meat, sweets, and sugar-containing beverages while increasing the intake of potassium, magnesium, and calcium but with variable restriction in dietary sodium.64,65 Both DASH diets reduced blood pressure within 4 weeks by 10/5 mm Hg or more, which is at least as effective as one antihypertensive medication. In the Nurses Health Study, adherence to the DASH dietary pattern was associated with a lower risk of CHD by 14% in those with the highest adherence to the diet. 66
Fats
The role of fats in CHD has been evaluated in 72 clinical trials with over 600,000 patients. 67 This included 32 observational studies of FA intake, 17 observational studies of FA biomarkers and 27 randomized clinical trials (RCTs) of FA supplements. Dietary TFA intake increased CHD by 16%, saturated fat intake increased CHD by 2% (worst with palmitic and stearic FA based on circulating FA biomarkers), omega 6 FA increased CHD by 1%, MUFA decreased CHD by 1% and omega 3 FA decreased CHD by 7%.67,68 Intakes of MUFAs and PUFAs are associated with a lower risk of CHD and death, whereas SFA and TFA intakes are associated with a higher risk of CHD. The replacement of SFAs with MUFAs and PUFAs or TFA with MUFAs was inversely associated with CHD.67–69
A large meta-analysis on omega 3 FA 70 reviewed 18 RCTs (93,000 subjects) and 16 prospective cohort studies (732,000 subjects) examining eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) from foods or supplements and CHD, including MI, sudden cardiac death, coronary death and angina in primary and secondary prevention. Among RCTs, there was a nonstatistically significant 6% reduction in CHD risk with EPA + DHA. Subgroup analyses of data from RCTs indicated a statistically 14–16% significant CHD risk reduction with EPA+DHA among higher-risk populations, including participants with elevated triglyceride levels over 150 mg/dl and elevated LDL-C above 130 mg/dl. Meta-analysis of data from prospective cohort studies resulted in a statistically 18% significant reduction of CHD for higher intakes of EPA + DHA > 1 g per day and risk of any CHD event. The sudden cardiac death (SCD) rate was reduced by 47%. For those with high TG over 150 mg/dl, doses of omega 3 FA > 1 g per day reduced CHD by 25%. These results and others indicate that EPA + DHA may be associated with a reduction in CHD risk, with a greater benefit observed among higher-risk populations in RCTs and those taking higher doses.
Saturated fatty acids
The relationship of CHD to SFA intake is controversial in published studies, with widely variable results.56,67,71–77 The long-chain FAs (LCFAs) which include C-12–C-18 (lauric, myristic, palmitic and stearic acids) show significant associations with CHD, but the short-chain FAs (SCFAs) which include butyric–capric do not.56,67,71–77 LCFAs induce insulin resistance, dysglycemia, metabolic syndrome, T2DM, dyslipidemia, obesity, thrombosis, endothelial dysfunction, CVAs, CHD and MI, whereas SCFAs do not cause any of these metabolic or medical problems. The replacement of SFA with an isocaloric intake of PUFA, omega 3 FAs, MUFA, a plant-based diet or whole grains reduces the risk of CHD, but an isocaloric replacement of SFA with trans fats (TFAs), omega 6 fatty acids, processed animal fat, refined carbohydrates, starches and high fructose corn syrup (HFCS) increases the risk of CHD.56,67,71–77
Coconut oil
In a meta-analysis of 21 studies with 8 clinical trials and 13 observational studies, coconut oil increased TC and LDL more than PUFA, increased HDL and increased TG with no change in the TC/HDL ratio. There was no change in CV events. 78 Coconut oil is 92% SFA, mostly lauric acid which acts mostly like an LCFA not a medium-chain fatty acid (MCFA) or medium-chain triglyceride (MCT), which is C-10 or less (direct portal vein absorption not via micelles and more water soluble). Only 4% of coconut oil is MCT of C-10 or less fatty acids.
Milk and milk products
Recent clinical studies indicate that milk, milk peptide and milk products reduce CHD, diabetes mellitus (DM), CVA and atherosclerosis.79,80 They improve insulin resistance, postprandial hyperglycemia, lower BP [increases NO and improves FMD and endothelial dysfunction (ED)] and decreases inflammation and oxidative stress.79,80
Carbohydrates and sugar substitutes
Sugars, refined carbohydrates, HFCS, starches and TFAs confer more risk for dyslipidemia and CHD than SFA. Omega 3 FAs, MUFAs, fermented foods, fiber, fruit + vegetables, dairy, TMD and the DASH 2 diet reduce CHD. 81 In a population-based cohort study of 39,786 participants over 18 years, daily diet soft drink consumption increased the risk of CVA by 21%. 82 The National Health Service (NHS) and Health Professioanls Follow-Up Study (HPFUS) showed both sugar-sweetened and low-calorie sodas significantly increased the risk of stroke by 16% per one serving per day and CHD by 20%. 83 Sugar substitutes increase the risk for obesity, weight gain, metabolic syndrome, T2DM and CHD. The sugar substitutes interfere with learned responses that normally contribute to glucose and energy homeostasis, kill the microbiome, alter leptin levels and decrease satiety.84,85
Vegetarian diets
Vegetarian diets significantly reduce CVD, CHD and CAC that is proportional to the dietary intake.86–89 In the EPIC Study of 44,561 participants in England and Scotland followed for 11.6 years, the body mass index (BMI), lipids and BP were all reduced in the vegetarian group and had a 32% lower incidence of CHD after adjustment for other CHD risk factors. 86 A study of 96,469 Seventh-day Adventist men and women from 2002 to 2007 demonstrated a 12% decrease in total mortality, 15% in vegans, 9% in lacto–ovo vegetarians, 19% in pesco–vegetarian individuals and 8% in semivegetarians, which was primarily related to decreases in CVD. 87 In a meta-analysis of nine cohort studies of 222,081 men and women, the overall reduction in CHD risk was 4% for each additional portion of fruit and vegetable intake per day (p = 0.0027) and 7% for each additional serving of fruit (p = 0.0001). 89 The association between vegetable intake and CHD risk was heterogeneous (p = 0.0043), more marked for CV mortality (0.74, p < 0.0001) than for fatal and nonfatal MI (0.95, p = 0.0058). 89 Dark green leafy vegetables had the most dramatic reduction in CHD risk. Some vegetarian diets may be deficient in many nutrients which require supplemental B12, vitamin D, omega 3 FA, iron, calcium, carnitine, zinc and some high quality amino acids and protein. 90 Other studies suggest many other problems, such as decreased sulfur amino acid intake with a low elemental sulfur, increased homocysteine and oxidative stress, lower cysteine (33% of control) and glutathione (63% of controls). In addition, lean muscle mass was 10% lower and there may be an increased risk of subclinical malnutrition and CVD. 90
Protein diets
Recent studies show either no correlation or an inverse correlation of grass-fed beef, wild game, organically fed animals and other sources of protein with CHD.91–96 The Paleolithic diet has also shown reductions in total mortality of 23% and CV mortality of 22% in a cohort study of 21,423 participants. 91 All meat (including red meat, fish, seafood, poultry) had an inverse relationship to CVD mortality in men in Asian countries. 93 Other meta-analyses showed no association between red meat consumption and CHD but found that processed red meat increased risk of hypertension, total mortality, CHD and DM risk.92–96 In the BOLD (Beef in an Optimal Lean Diet Study) trial, a low SFA, heart-healthy diet that contains lean beef elicits a favorable effect on CVD, lipids and lipoprotein risk factors that are comparable with the DASH diet. 92 This may be related to certain amino acids in meat versus vegetables, such as the lysine and arginine content.
Specific dietary and nutritional components
Several dietary and nutritional components have been shown to interrupt the inflammatory vascular receptors such as PRRs, NLRs and TLRs. 20 These include:
Curcurmin (tumeric) blocks TLR 4, nucleotide-binding oligomerization domain (NOD) 1, and NOD 2;
Cinnamaldehyde (cinnamon) blocks TLR 4;
Sulforaphane (broccoli) blocks TLR 4;
Resveratrol (nutritional supplement, red wine, grapes) blocks TLR 1;
Epigallocatechin gallate (EGCG) (green tea) blocks TLR 1;
Luteolin (celery, green pepper, rosemary, carrots, oregano, oranges, olives) blocks TLR 1;
Quercetin (tea, apples, onion, tomatoes, capers) blocks TLR 1.
A prospective study of 42 participants over 2 years showed a significant reduction in progression of CHD as assessed by electron beam tomography CAC compared with historical controls using a phytonutrient concentrate with a high content of fruit and vegetable extracts. The CAC score increased by 19.6% in treated patients versus 34.7% in controls (p < 0.009). 97
Caffeine
The cytochrome P450-CYP1A2 genotype modifies the association between caffeinated coffee intake and the risk of hypertension, CVD, CHD and MI in a linear relationship.98–105 Caffeine is exclusively metabolized by CYP1A2 to paraxanthine, theobromine and theophylline. 98 The gene lies on chromosome 15q24.1 and the single nucleotide polymorphism (SNP) is rs7762551 A to C. 98 The C SNP decreases enzymatic activity. 98 Caffeine also blocks vasodilating adenosine receptors. 105 The rapid metabolizers of caffeinated coffee IA/IA allele have lower BP and lower risk of MI. 103 Hypertension has a 0.36–0.80 relative risk ratio, with an average reduction in BP of 10/7 mm Hg. MI shows a 17–52% reduction. This SNP represents about 40–45% of the population.98–103 The slow metabolizers of caffeine IF/IF or IA/IF allele have higher BP 8.1/5.7 mm Hg, lasting >3 h after consumption, tachycardia, increased aortic stiffness, higher pulse wave velocity, vascular inflammation and increased catecholamines.98,100 Hypertension risk is increased [1.72–3.00 relative risk (RR)]. 99 Based on age and consumption, the risk of MI will vary. At age 59 there was a 36% increase in MI with 2–3 cups/day and a 64% increase with 4 cups/day or more. Under the age of 59, MI increased by 24% (1 cup/day), 67% (2 cups/day) and 233% (4 or more cups/day).103,104 This SNP represents about 55–60% of the population.
Nutritional supplements
Numerous nutritional supplements have demonstrated improvement in surrogate endpoints (BP, lipids, glucose, carotid IMT, coronary calcification, etc.). However, there are limited data that nutraceutical supplements reduced hard CV endpoints related to CHD, MI, and CHF. The nutritional supplements that will be discussed below in the paper offer some evidence of surrogate endpoints for CHD and others indicate some improvement in angina, CHD, MI and CHF. Only those supplements with scientific data in these mentioned CV outcomes will be reviewed in this paper. The dose of the supplement will vary depending on prevention or treatment considerations, age, body weight and concomitant medications or other supplements. All nutritional supplements should be obtained from highly certified and reputable nutritional supplement companies that undergo scrutiny with state and federal authorities to assure safety and efficacy standards.
Omega 3 fatty acids
Omega 3 FA supplementation reduces all-cause mortality and MI in primary and secondary prevention trials, as well as many other CV outcomes70,106–120 Omega 3 FAs decrease MI and CHD 18% more with concomitant use of statins, 110 reduce stent restenosis, 108 CABG occlusion,112,113 plaque formation,114,115 coronary artery calcification,114,115 atherosclerosis,114,115 improve the lipid profile,18,116 lower glucose and improve insulin resistance117–119 and reduce BP.2,4,16,120 The dose prescribed will depend on the condition being treated, as well as age, body weight and use of concomitant medications and other nutritional supplements. It is best to use a balanced formulation with DHA, EPA, gamma linolenic acid (GLA) and gamma–delta tocopherols. This will prevent oxidation in the cell membranes and reduce depletions of the EPA and DHA by GLA or vice versa.18,116,120
D-Ribose
D-Ribose improves cardiomyopathy, systolic and diastolic CHF, acute and chronic CHD and angina, stabilizes and energizes the heart post MI and improves the postoperative ejection fraction in CABG. D-Ribose increases myocardial adenosine triphosphate (ATP) and energetics within 1 h of administration and lasts for about 6–8 h. It is well tolerated, with minor side effects including diarrhea and rarely mild reductions in serum glucose. The dose is 5 g three times daily.121–126
Vitamin K
In an NHS cohort of 72,874 female nurses, there was a 16% RR reduction in CHD from lowest to highest quintile of vitamin K1 (phylloquinone) intake. 127 In the HPFUS of 40,087 men, there was a 13–16% RR reduction in CHD. 128 The Multi-Ethnic Study of Atherosclerosis (MESA) showed that a low vitamin K1 dietary intake showed increased CAC, especially in those on antihypertensive drugs, by twofold. 129 A population-based study of 4807 participants found the incident risk for CHD was reduced by Vitamin K1 and Vitamin K2 (menaquinone). K2 reduced CHD by 57% in the upper versus lower tertile and K2 reduced all-cause mortality by 26% in the upper versus lower tertile. K2 reduced aortic calcification by 52% in the upper versus lower tertile and reduced total mortality by 26%, but there was no association with K1. 130 The vitamin-K-dependent matrix Gla protein is a potent inhibitor of the arterial calcification and may become a noninvasive biochemical marker for vascular calcification. Vitamin K2 is considered more important for vascular system health, if compared with vitamin K1. 131 The recommended dose of K2 MK7 is at least 100–150 mg per day.
Carnitine
L-carnitine reduced the all-cause mortality by 27%, ventricular arrhythmias by 65% and angina by 40% following an acute MI compared with placebo in 13 controlled trials of 3629 participants. 132 There was no change in CHF or recurrent MI. Concerns about the possible effects of carnitine on the microbiome and TMAO (trimethylamine N-oxide) production have to be evaluated against the positive effects in these studies. It is not clear if TMAO has a cause-and-effect relationship with CHD. 133 Carnitine is important in the transport of LCFAs of C-12 or greater into the myocardium for beta oxidation of FAs that supply 60% of the ATP for the cardiac myocyte. 134
Curcumin
In a study of 121 patients, curcumin reduced MI post CABG from 30% to 13% (p < 0.038) at 4 g per day given 3 days before and 5 days after CABG. 135 The hsCRP, malondialdehyde and N-terminal prohormone of B-type brain natriuretic peptide (NT-pro BNP) were also lower. Curcumin significantly attenuated collagen deposition in mice after coronary artery ligation-induced MI and inhibited cardiac fibroblast proliferation and migration and metalloproteinase expression. In addition, there was downregulation of SIRT1 (Sirtuin 1) after MI that was attenuated by curcumin pretreatment, which indicated that the activation of SIRT1 might be involved in the protective action of curcumin. 136
Co-enzyme Q10
Co-enzyme Q10 (CoQ10) reduces post-MI reperfusion ventricular arrhythmias, improved LV function and total cardiac death.137,138 In a double blind placebo controlled (DBRPC) trial of 144 subjects with acute MI, CoQ10 at 120 mg per day administered within the first 3 days of an MI resulted in significant improvements in the treated group in all parameters (p < 0.05): 138
Angina (9.5% versus 25.3%);
Arrhythmias (9.5% versus 25.3%);
LVF improved (8.2 versus 22.5%);
Total cardiac events and death reduced at 15 versus 30.9% (p < 0.02).
The Q –SYMBIO Trial of CoQ10 and CHF is the most important of all the studies yet published on CoQ10 and heart failure. 139 This was a randomized DBRPC trial of 420 patients with CHF New York Heart Association (NYHA) class III and IV over 10 years. Subjects were administered 2 mg/kg CoQ10 per day (100 mg tid) versus placebo plus standard therapy. Serum levels of CoQ10 increased three times above baseline. The primary short-term endpoints were NYHA function class, a 6 min walk test, NT-pro BNP and ejection fraction (EF). There was no difference between groups and nonsignificant change in EF in these short-term primary endpoints. The CoQ10-treated subjects had reduced major adverse cardiac events by 50% (p = 0.003, CI: 32–80) and all-cause mortality by 42%. Major adverse CV events were defined as hospitalization or death due to CHF, CV/MI death, SCD, cardiac transplant or mechanical circulatory support. There was a reduction in CV mortality from 16% to 9% (p = 0.026), all-cause mortality from 18% to 10% (p = 0.018) and reduction in the incidence of hospital stays for CHF (p = 0.033). The NYHA class was significantly improved in the CoQ10 group at 2 years (p = 0.028).
Conclusion
The top five CV risk factors, as presently defined, are not an adequate explanation for the current limited reduction in CHD or the ‘CHD gap’. Proper definition and analysis of the top five risk factors, evaluating the other 395 risk factors and downstream mediators should be included with measurement of the three finite responses of inflammation, oxidative stress and vascular immune dysfunction, micronutrient testing, CV genetics, nutrigenomics, metabolomics, gene expression testing and noninvasive vascular testing. Early detection coupled with aggressive prevention and treatment of all CV risk factors will diminish the progression of functional CV abnormalities, CV structural problems and clinical CVD. If we wish to revolutionize the prevention and treatment of CVD then a new approach should be implemented in the clinical setting. This will be achievable by using a combination of targeted, personalized and precision treatments with optimal nutrition and nutraceutical supplements coupled with optimal exercise, weight and body composition management and a reduction in tobacco use. Approximately 80% of CHD can be prevented with this approach.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author declares that there is no conflict of interest.