
Abstract
Endothelial dysfunction underlies multiple cardiovascular consequences of chronic kidney disease (CKD) and antecedent diabetes or hypertension. Endothelial insults in CKD or end-stage renal disease (ESRD) patients include uremic toxins, serum uric acid, hyperphosphatemia, reactive oxygen species, and advanced glycation endproducts (AGEs). Sevelamer carbonate, a calcium-free intestinally nonabsorbed polymer, is approved for hyperphosphatemic dialysis patients in the US and hyperphosphatemic stage 3–5 CKD patients in many other countries. Sevelamer has been observed investigationally to reduce absorption of AGEs, bacterial toxins, and bile acids, suggesting that it may reduce inflammatory, oxidative, and atherogenic stimuli in addition to its on-label action of lowering serum phosphate. Some studies also suggest that noncalcium binders may contribute less to vascular calcification than calcium-based binders. Exploratory sevelamer carbonate use in patients with stages 2–4 diabetic CKD significantly reduced HbA1c, AGEs, fibroblast growth factor (FGF)-23, and total and low-density lipoprotein (LDL) cholesterol versus calcium carbonate; inflammatory markers decreased and defenses against AGEs increased. Sevelamer has also been observed to reduce circulating FGF-23, potentially reducing risk of left ventricular hypertrophy. Sevelamer but not calcium-based binders in exploratory studies increases flow-mediated vasodilation, a marker of improved endothelial function, in patients with CKD. In contrast, lanthanum carbonate and calcium carbonate effects on FMV did not differ in hemodialysis recipients. The recent INDEPENDENT-CKD randomized trial compared sevelamer versus calcium carbonate in predialysis CKD patients (investigational in the US, on-label in European participants); sevelamer reduced 36-month mortality and the composite endpoint of mortality or dialysis inception. Similarly, INDEPENDENT-HD in incident dialysis patients showed improved survival with 24 months of sevelamer versus calcium-based binders. This review discusses recent exploratory evidence for pleiotropic effects of sevelamer on endothelial function in CKD or ESRD. Endothelial effects of sevelamer may contribute mechanistically to the improved survival observed in some studies of CKD and ESRD patients.
Keywords
Introduction
Chronic kidney disease (CKD) multiplies cardiovascular risk for affected patients even beyond the risk conferred by antecedent diabetes and/or hypertension. Patients with CKD stage 5 have 10–20 times the general public’s risk of cardiovascular death after stratification for age, gender, and ethnicity (Figure 1a) [Foley et al. 1998] and are more likely to die of cardiovascular disease than to progress to dialysis. In fact, cardiovascular disease is the most common cause of death for patients with CKD or end-stage renal disease (ESRD). This risk stems from multiple pathogenic processes affecting the heart and vessels, e.g. atherogenic [Stenvinkel et al. 2003], inflammatory, and thrombogenic states; endothelial dysfunction; and disrupted blood pressure regulatory molecules [Kovesdy and Kalantar-Zadeh, 2008]. In addition, reduced vitamin D receptor activation, which occurs in CKD mineral and bone disorder (MBD), is associated with hypertension, left ventricular hypertrophy, vascular stiffening, and/or accelerated atherosclerosis and arteriosclerosis, all of which contribute to cardiovascular mortality [Kovesdy and Kalantar-Zadeh, 2008].

Increased cardiovascular mortality risk associated with Stage 5 CKD versus the general population. Reproduced with kind permission from Elsevier (Foley et al. 1998).
Vascular calcification is a major source of ESRD-related cardiovascular risk. It results from metabolic, inflammatory, and developmental effects of CKD (especially CKD-MBD) upon vascular cells and structures [Sage et al. 2010]. While classic CKD-related vascular calcification affects the smooth muscle layer (tunica media), hyperphosphatemia and calcium loading may also worsen calcification of atherosclerotic plaques in the endothelium (intima or neointima). Vascular calcification prevalent at dialysis initiation can progress rapidly, contributing to the high mortality risk of the first 90 days on dialysis [Block et al. 2007]. Vascular calcification also predicts mortality risk in prevalent dialysis patients. In a maintenance hemodialysis patient cohort, high overall and vessel-specific coronary artery calcification scores (101–400 or >400) predicted significantly higher 6-year mortality risk versus patients with zero calcification scores at baseline [Shantouf et al. 2010].
Phosphate binder treatment (approved for hyperphosphatemic dialysis patients in the US and hyperphosphatemic stage 3–5 CKD patients in many other countries) reduces cardiovascular risk in renal disease. Phosphate loading has deleterious effects on vascular smooth muscle (tunica media) and endothelium; it contributes to cardiovascular calcification and atherogenesis both in patients with CKD-MBD and the general population [Ellam and Chico, 2012]. In incident dialysis patients, use of phosphate binders within the first 90 days of dialysis has been observed in the following study to reduce early dialytic mortality versus patients not receiving binders [Isakova et al. 2009]. In the extension of the Renagel in New Dialysis (RIND) randomized open-label study of 129 incident hemodialysis patients, baseline vascular calcification (e.g. coronary artery calcification score) and selected phosphate binders independently predicted mortality on 44-month follow up [Block et al. 2007]. Post hoc analysis of the Cholesterol and Recurrent Events (CARE) study [Tonelli et al. 2005] of patients with prior myocardial infarctions and hyperlipidemia, including those with normal glomerular filtration rate (GFR), suggested that increasing serum phosphate even within the normal range may also be associated with cardiovascular events in renally normal persons. The CARE authors state that ‘When participants were divided into 4 categories based on their baseline phosphate level (<2.5, 2.5 to 3.4, 3.5 to 3.9, and ≥4 mg/dL), a graded relation between phosphate and death was observed after adjustment for age, race, and sex (p for trend = 0.01)’ [Tonelli et al. 2005]. Finally, the INDEPENDENT-HD study [Di Iorio et al. 2013] of 466 incident dialysis patients receiving sevelamer or calcium-based binders for 24 months showed that sevelamer recipients had significantly lower arrhythmic, all-cardiovascular, and all-cause mortality by month 36 than calcium-binder recipients.
Sevelamer (Figure 2) is a metal-free, nonabsorbed polymeric anion exchange resin phosphate binder. In addition to phosphate, it binds other substances in the gut (e.g. bile acids, bacterial endotoxins, and advanced glycation endproducts [AGEs]), leading to pleiotropic effects specific to a polymeric phosphate binder [Charmot, 2012]. Two formulations exist: sevelamer hydrochloride and sevelamer carbonate. Sevelamer carbonate has been shown to lower serum phosphate to the same extent as sevelamer hydrochloride; however, whereas sevelamer hydrochloride has been reported to decrease serum bicarbonate levels and contribute to metabolic acidosis in dialysis recipients, sevelamer carbonate is associated with higher bicarbonate levels and less acidosis [Fan et al. 2009; Pai and Shepler, 2009; Savica et al. 2008]. The polymeric nature of sevelamer and its ability to bind not only phosphate but also other molecules in the intestine drive its pleiotropic effects. Observed reduction of early dialytic cardiovascular mortality with sevelamer hydrochloride may have several possible mechanisms: not only lowering of serum phosphate, lipids, and reduced vascular calcification but potentially also anti-inflammatory and anti-uremic effects [Evenepoel, 2007]. Phosphate-reducing and possible phosphate-independent biochemical interactions of sevelamer are summarized in Figure 3.

Structure of sevelamer.

Summary of proposed phosphate-reducing and phosphate-independent effects of sevelamer.
Several studies (Table 1) suggest that sevelamer may be associated with less progression of coronary artery and aortic calcifications than calcium-based binders in dialysis patients, for whom it is indicated in the US [Asmus et al. 2005; Block et al. 2005; Braun et al. 2004; Chertow et al. 2002, 2003; Kakuta et al. 2011; Shantouf et al. 2010] and hyperphosphatemic patients with CKD not requiring dialysis, for whom it is also indicated under European Medicines Agency approval [Block et al. 2012; Caglar et al. 2008; Di Iorio et al. 2012; Russo et al. 2007; Yilmaz et al. 2010]. Incident dialysis patients assigned to calcium-based binder use in the 18-month parent RIND study showed progression of vascular calcification [Block et al. 2005] and increased 44-month mortality in the RIND extension [Block et al. 2007], whereas assignment to sevelamer hydrochloride use was associated with attenuated 18-month progression of vascular calcification [Block et al. 2005] and decreased 44-month mortality (Figure 4) [Block et al. 2007]. The metallic noncalcium phosphate binder lanthanum carbonate similarly was associated with less progression of coronary calcification than calcium carbonate in a pilot study of hemodialysis patients [Kalil et al. 2012]. Authors of another pilot randomized controlled trial in 45 dialysis patients concluded that ‘Lanthanum carbonate was associated with reduced progression of aortic calcification compared with CC [calcium carbonate] in HD patients over 18 months’ [Toussaint et al. 2011].
Effects of sevelamer on vascular calcification in patients with CKD requiring or not requiring dialysis.
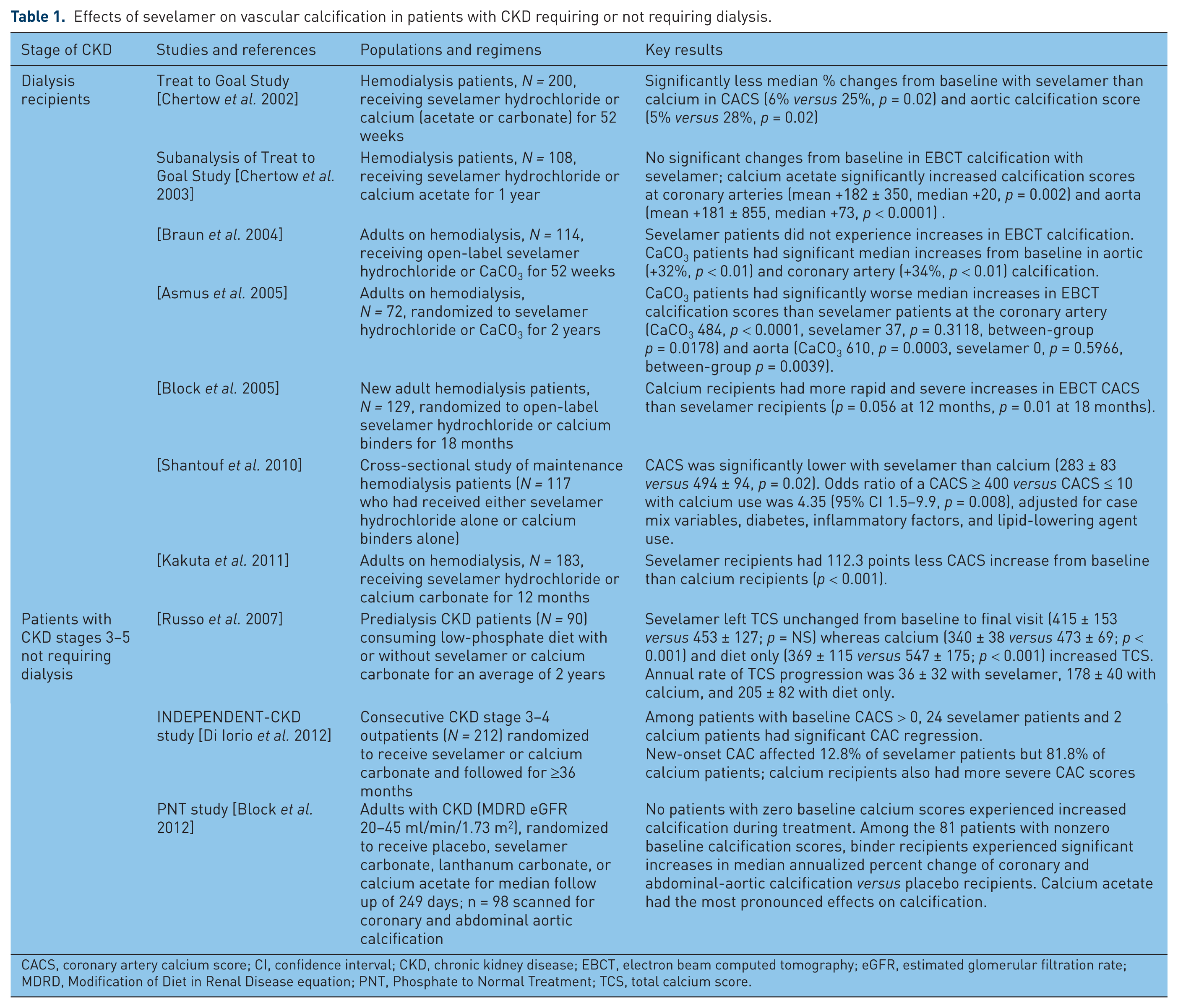
CACS, coronary artery calcium score; CI, confidence interval; CKD, chronic kidney disease; EBCT, electron beam computed tomography; eGFR, estimated glomerular filtration rate; MDRD, Modification of Diet in Renal Disease equation; PNT, Phosphate to Normal Treatment; TCS, total calcium score.

Effects of sevelamer versus calcium on mortality in patients new to hemodialysis. Adapted with kind permission from Macmillan Publishers Ltd. (Block et al. 2007).
The vascular endothelium is a key mediator of vascular homeostasis [Santoro et al. 2010]. It responds to mechanical stimuli from blood flow and to blood-borne metabolic and endocrine signals; in turn it sends chemical signals to regulate vascular tone, permeability, proliferation, coagulation, and inflammation [Santoro et al. 2010]. Endothelial inflammation is one of the initiating events of atherosclerosis [Ross, 1999]. Endothelial pathology, including the adverse effects of CKD- and ESRD-related biochemical derangements [Santoro et al. 2010], links comorbidities and risk factors to many cardiovascular disorders. Hypertension, hyperlipidemia, hyperuricemia, menopause, diabetes, and resulting AGEs (e.g. indoxylsulfate), and smoking and resulting reactive oxygen species contribute to dysfunction and damage in endothelial and vascular smooth muscle cells. Endothelial dysfunction in turn contributes to vascular pathologic processes: vasoconstriction, apoptosis, lipid deposition, leukocyte adhesion, vascular smooth muscle cell growth, and thrombosis. [Pool and Taylor, 2007]
Objective
This review will discuss recent evidence for effects of sevelamer on parameters affecting vascular endothelial function.
Normal endothelial function
The vascular endothelium, a monolayer of endothelial cells weighing 1.5 kg with surface area equivalent to four tennis courts, acts as the main integrator of vascular homeostasis [Santoro et al. 2010]. It senses mechanical stimuli (pressure and shear stress) related to blood flow dynamics and hormonal stimuli related to vasomotor regulation [Santoro et al. 2010]. In turn, it releases biochemical mediators to regulate vascular tone, coagulation, vascular permeability, and inflammation [Santoro et al. 2010].
Endothelial dysfunction in CKD, hypertension, and diabetes
Endothelial dysfunction is an important common link among hypertension, diabetes, hypertensive or diabetic CKD, and their cardiovascular sequelae. Cardiovascular disorders such as atherosclerosis [Ross, 1999], peripheral and coronary artery disease, and chronic heart failure both contribute to and are worsened by endothelial dysfunction. Vascular consequences of endothelial dysfunction include impaired response to shear stress (e.g. reduced flow-mediated vasodilation [FMV]), reduced vascular compliance, inflammation, prothrombic state, increased cellular permeability, and cell proliferation [Santoro et al. 2010]. Potential sources of endothelial dysfunction in CKD include uremic toxins, serum phosphate, serum uric acid, oxidative stress, and AGEs; thus patients with CKD are likely to experience additional endothelial insults beyond the effects of hypertension or diabetes [Pool and Taylor, 2007].
In patients with CKD, endothelial dysfunction probably contributes to both progression of renal disease and cardiovascular sequelae. Treatments intended to protect the endothelium, improve endothelial function, and prevent atherosclerosis are commonly prescribed [Turner et al. 2012], such as inhibitors of the renin–angiotensin system, other antihypertensives, insulin-sensitizing agents, lipid-lowering agents to reduce LDL-mediated inflammation, l-arginine, and interventions against oxidative stress (e.g. smoking cessation, antioxidant consumption, and dietary AGE reduction). Acute increases of serum phosphate (e.g. postprandial phosphate absorption peaks) have been observed to stimulate endothelial dysfunction by increasing reactive oxygen species and decreasing vasodilatory nitric oxide production [Shuto et al. 2009]. High serum phosphate levels also may contribute to atherogenesis with or without intimal calcification, and may exert direct endothelial toxicity [Ellam and Chico, 2012]. Conversely, phosphate binding with sevelamer but not calcium-based binders has been shown in exploratory studies to increase FMV, a marker of improved endothelial function, in patients with CKD [Caglar et al. 2008; Yilmaz et al. 2012]. In contrast, lanthanum carbonate and calcium carbonate did not differ in their effect on FMV in hemodialysis recipients [Kalil et al. 2012].
Pleiotropic effects of sevelamer affecting endothelial function and cardiovascular risk
In addition to lowering serum phosphate and reducing calcium burden, sevelamer hydrochloride or carbonate exerts diverse pleiotropic effects on parameters associated with cardiovascular risk in CKD/ESRD [Evenepoel, 2007; Nikolov et al. 2006] many of which may be mediated by their relationships to endothelial function. Specific effects and individual studies are summarized in Table 2. Broad categories of effects include direct improvements in endothelial and/or vascular function [Brandenburg et al. 2009, 2010; Caglar et al. 2008; Takenaka and Suzuki, 2005; Yilmaz et al. 2012]; reduced progression of vascular calcification in patients with dialysis-dependent ESRD [Asmus et al. 2005; Block et al. 2005; Braun et al. 2004; Chertow et al. 2002, 2003; Kakuta et al. 2011; Shantouf et al. 2010] or predialysis CKD [Caglar et al. 2008; Di Iorio et al. 2012; Russo et al. 2007]; reduced absorption of bile acids from the gut [Braunlin et al. 2002], leading to lower serum total and LDL cholesterol [Bleyer et al. 1999; Di Iorio et al. 2012; Evenepoel et al. 2009; Ferramosca et al. 2005; Gulati et al. 2010; Hervas et al. 2003; Lin et al. 2011], reduced absorption of endotoxin from gut bacteria [Stinghen et al. 2010; Sun et al. 2009], lower levels of inflammatory mediators and biomarkers [Caglar et al. 2008; Ferramosca et al. 2005; Shantouf et al. 2008, 2010; Stinghen et al. 2010; Sun et al. 2009; Yamada et al. 2005], reduced absorption of dietary AGEs [Vlassara et al. 2012a], and decreased carotid intima-media thickness (CIMT), a surrogate marker of atherosclerosis [Boaz et al. 2011]. CKD fosters pro-atherogenic conditions through multiple pathways, including oxidative stress, increased infection and inflammation, decreased clearance of inflammatory mediators, increased formation and decreased clearance of AGEs, LDL oxidation, and adverse changes in endothelial and vascular smooth muscle cells [Stenvinkel et al. 2003]. Atherogenesis and inflammation, not only phosphate–calcium metabolism, in turn contribute to vascular calcification [Sage et al. 2010]. Sevelamer thus may ameliorate multiple processes that contribute to calcification and other sources of cardiovascular disease risk in CKD (Figure 5).
Summary of pleiotropic effects of sevelamer versus calcium-based binders (additional to lowering of serum phosphate and attenuation of vascular calcification) on multiple parameters affecting cardiovascular risk in CKD/ESRD.

AGE, advanced glycation endproduct; BSA, bovine serum albumin; CAC(S), coronary artery calcification (score); CI, confidence interval; CKD, chronic kidney disease; CT, computed tomography; EBCT, electron beam computed tomography; eGFR, estimated glomerular filtration rate; FGF-23, fibroblast growth factor-23; FMV, flow-mediated vasodilation; HDL, high-density lipoprotein; hsCRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; MDRD, Modification of Diet in Renal Disease equation; OR, odds ratio; PTH, parathyroid hormone; PWV, pulse wave velocity; QCT, quantitative computed tomography; TCS, total calcium score.

Potential pleiotropic effects of sevelamer affecting parameters involved in the inflammatory, metabolic, or developmental modulation of vascular calcification. Factors that have been shown to respond to sevelamer are shown in yellow ovals. Adapted with kind permission from Macmillan Publishers Ltd. (Sage et al. 2010).
Endothelium-dependent vasodilation and its determinants
Hyperphosphatemia may directly induce endothelial dysfunction and damage, as indicated by in vitro, animal, and human studies [Ellam and Chico, 2012]. In healthy human volunteers, acute postprandial increases in serum phosphate after a breakfast containing 1200 mg phosphate decreased nitric oxide synthase activity and FMV [Shuto et al. 2009].
Circulating fetuin-A, the main inhibitor of soft-tissue calcification, decreases as GFR declines, parallel to an increase in the vasoconstrictive protein endothelin-1 [Cottone et al. 2010]. Decreased fetuin-A is correlated not only with vascular calcification itself [Sage et al. 2010] but also with endothelial vasomotor dysfunction shown by decreased FMV [Caglar et al. 2008]. A short-term study showed FMV and circulating fetuin-A to decrease significantly in patients with stage 4 nondiabetic CKD compared with healthy controls; 8 weeks of sevelamer treatment significantly increased fetuin-A and FMV in this study’s CKD participants [Caglar et al. 2008]. FMV and fetuin-A levels had independent multivariate association both at baseline and after treatment. In another 8-week randomized investigation of 100 nondiabetic patients with stage 4 CKD [Yilmaz et al. 2012], sevelamer significantly increased brachial artery endothelium-dependent FMV. Changes in FMV were significantly associated with changes in fibroblast growth factor-23 (FGF-23), phosphate, parathyroid hormone (PTH), fetuin-A, and C-reactive protein (CRP); phosphate and PTH lost their predictive power on multivariate analysis. Fetuin-A increases also have been associated with significant reductions of all-cause and cardiovascular mortality in dialysis patients [Hermans et al. 2007].
A possible challenge to the postulated interrelationship of endothelial dysfunction (e.g. impaired FMV) and vascular calcification or atherosclerosis comes from a 6-month pilot randomized study of lanthanum carbonate in hemodialysis patients [Kalil et al. 2012]. Lanthanum carbonate recipients experienced significantly less progression of coronary artery calcification than did patients receiving other binders (mostly combinations of sevelamer and calcium binders); however, FMV and inflammatory markers did not differ between treatments. The study authors stated that this was the first study to dissociate FMV changes from progression of coronary artery calcification [Kalil et al. 2012].
Advanced glycation endproducts
AGEs are highly reactive, oxidative [Kakuta et al. 2011; Vlassara et al. 2012b], inflammatory, and atherogenic [Prasad et al. 2012] molecules formed from oxidation of carbohydrates, lipids, and amino acids [Uribarri and Tuttle, 2006]. AGEs are produced during normal glucose oxidation and immune-cell function [Uribarri and Tuttle, 2006], in nonenzymatic sugar–protein reactions [Prasad et al. 2012], and during dry-heat cooking of foods [Uribarri and Tuttle, 2006]. In the body AGEs form adducts with structural proteins, stiffening cardiovascular structures and altering their mechanics [Falcao-Pires et al. 2012; Prasad et al. 2012]. AGEs accumulate with declining eGFR in patients with CKD, induce expression of the inflammatory Receptor for AGEs (RAGE) on neutrophils, and impair endothelial vasomotor function [Linden et al. 2008]. Plasma levels of the AGE pentosidine are correlated with cardiovascular calcification in patients with diabetic CKD [Kakuta et al. 2011] and with CIMT, a direct measure of atherosclerosis, in first-year dialysis patients [Suliman et al. 2006]. A low-AGE diet reduced circulating AGEs significantly in a study of healthy subjects and predialysis CKD patients, showing that intestinal AGE absorption determines plasma AGE concentrations [Vlassara et al. 2009]. Sevelamer reduced serum and intracellular AGEs (likely through intestinal absorption of dietary AGEs) in patients with predialysis diabetic nephropathy [Vlassara et al. 2012b] and in diabetic dialysis patients [Vlassara et al. 2012a], and reduced circulating pentosidine in dialysis patients, whereas pentosidine was increased by calcium-based binders [Kakuta et al. 2011].
Vlassara and colleagues [Vlassara et al. 2012b] enrolled 20 patients with proteinuric diabetic CKD into a 2-month, open-label, crossover study comparing effects of sevelamer carbonate (1600 mg thrice daily with meals) versus calcium carbonate (1200 mg thrice daily with meals) on AGEs. Binder order was randomly assigned; patients received each binder for 8 weeks separated by a 1-week washout. Serum phosphate and calcium did not change from baseline on either binder in this brief study; 24-hour urinary phosphate excretion was decreased similarly by sevelamer and calcium. Blood glucose, dietary AGE intake, adipokines, and CRP remained similar between groups. Sevelamer (in comparison with calcium) reduced HbA1c, total cholesterol, triglycerides, 8-isoprostanes, and proteins modified with carboxymethyllysine (CML) or methylglyoxal. Calcium carbonate significantly increased 8-isoprostanes. In polymorphonuclear neutrophils, sevelamer carbonate but not calcium carbonate decreased tumor necrosis factor-α levels and increased messenger RNAs for AGE receptor 1 and sirtuin 1 (thus demonstrating reduced inflammatory responses and enhanced defenses against AGE-induced damage).
In 132 diabetic hemodialysis patients, sevelamer at 4.8 or 7.2 g/day for 3 weeks significantly reduced serum MG and CML from baseline and versus placebo, while sevelamer at 2.4 g/day did not change MG and CML from baseline. Vlassara and colleagues state that ‘An important therapeutic issue pointed out by this study was that sevelamer carbonate at a dosage of 2.4 g/day did not significantly reduce cytopathic AGEs. Because cytopathic AGEs are inducers of ROS and/or inflammation, assuring that patients reach and maintain the 4.8 g/day dosage level may represent an important therapeutic goal.’ [Vlassara et al. 2012a]
Fibroblast growth factor-23
Circulating FGF-23 increases progressively with declining renal function in CKD and ESRD in an effort to increase per-nephron phosphorus excretion [Slatopolsky, 2011]. Elevation of FGF-23 directly induces left ventricular hypertrophy [Faul et al. 2011] and is linked to endothelial/vascular dysfunction (impairment of FMV) in patients with CKD [Yilmaz et al. 2010, 2012] and community-dwelling elders in the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) [Mirza et al. 2009b]. High serum FGF-23 also predicted total body atherosclerosis scores in PIVUS [Mirza et al. 2009a] and carotid atherosclerosis scores in ESRD patients [Balci et al. 2010]. Circulating FGF-23 is negatively associated with circulating fetuin-A [Coen et al. 2011]; thus, increased FGF-23 may also contribute to endothelial dysfunction and vascular calcification by lowering fetuin-A levels. Calcium-based phosphate binders elevate serum FGF-23 in patients with CKD or ESRD, while noncalcium binders (sevelamer or lanthanum) reduce it [Cancela et al. 2011; Oliveira et al. 2010; Yilmaz et al. 2012]. In the Vlassara and colleagues diabetic nephropathy study [Vlassara et al. 2012b], sevelamer significantly decreased FGF-23 versus calcium in the subset of patients with baseline FGF-23 >70 pg/ml; overall, FGF-23 numerically decreased with sevelamer and numerically increased with calcium; although within-treatment changes from baseline were not statistically significant, the difference between treatments was significant (p = 0.04).
Inflammation
Endothelial and vascular inflammation are implicated in multiple cardiovascular disorders in the general population and patients with CKD/ESRD [Cottone et al. 2007, 2008; Nikolov et al. 2006; Ross, 1999; Santoro et al. 2010]. Sources of inflammation include oxidized LDL cholesterol, bacterial endotoxins (from gut absorption or infections of dialysis accesses or peripheral tissues [Hauser et al. 2011]), AGEs, and uremic toxins. Reactive oxygen species in turn are increased by inflammatory cell activity, leading to oxidative damage. Sevelamer has been shown to reduce circulating high-sensitivity C-reactive protein (hsCRP) in patients with CKD [Yilmaz et al. 2012] and on dialysis [Shantouf et al. 2008; Yamada et al. 2005], whereas calcium-based binders do not [Shantouf et al. 2008; Yilmaz et al. 2012]. In a short-term comparison of lanthanum carbonate and calcium carbonate in hemodialysis patients, inflammatory markers did not change in either group [Kalil et al. 2012]. Reduced levels of plasma endotoxins (reflecting reduced absorption from gut bacteria) were associated with sevelamer use in a pilot study of hemodialysis patients [Sun et al. 2009] and a prospective study of dialysis patients switched from calcium carbonate to sevelamer for 6 months of follow up [Stinghen et al. 2010]; in the latter cohort, hsCRP decreased in parallel to reduced endotoxemia. Animal studies in non-CKD mice suggest that lanthanum ions may reduce inflammatory mediator generation and secretion by the liver and macrophages (tumor necrosis factor-α and interleukin-β) in response to bacterial lipopolysaccharide [Guo et al. 2010, 2011].
Systemic inflammation in dialysis recipients can exacerbate anemia and impair response to anti-anemic agents; thus the effect of sevelamer (potentially reducing inflammatory stimuli) on anemia management is being evaluated. Preliminary data in 45 hemodialysis patients [Ikee et al. 2012] showed that sevelamer dose independently predicted responsiveness to erythropoiesis-stimulating agents (exemplified by reduced ‘ESA resistance index’, weekly erythropoietin dose divided by hemoglobin value); the authors called for further research into the effects of sevelamer on ESA responses.
Atherogenesis
CIMT, a surrogate marker of developing atherosclerosis [Boaz et al. 2011], is increased by inflammatory and oxidative damage [Drueke et al. 2002] and independently predicts cardiovascular events in patients with and without CKD [Drueke et al. 2002]. CIMT responds to many biochemical perturbations occurring in CKD. In hemodialysis patients, serum FGF-23 and CIMT were significantly correlated (r = 0.497, p = 0.0001), and elevated log FGF-23 independently predicted CIMT [Balci et al. 2010]. In another study, plasma pentosidine correlated with CIMT increases during the first dialysis year [Suliman et al. 2006]. Advanced protein oxidation products were associated with CIMT increases in hemodialysis patients receiving intravenous iron [Drueke et al. 2002]. Sevelamer exposure in dialysis patients was associated with significantly reduced CIMT in a nested cross-sectional substudy of the Sevelamer hydrochloride and Ultrasound-Measured femoral and carotid intima Media thickness progression in End-stage Renal disease (SUMMER) trial [Boaz et al. 2011]. The association of reduced CIMT with sevelamer use persisted after controlling for serum calcium, cardiovascular history, and body weight. Importantly, SUMMER analyzed patients with sevelamer exposure regardless of whether they also had received calcium binders.
In apolipoprotein E-deficient mice with chronic renal failure (CRF), both sevelamer hydrochloride and lanthanum carbonate reduced the progression of both atherosclerosis and vascular calcification (VC), and reduced vascular plaque expression of Type 1 collagen [Nikolov et al. 2012]. Sevelamer additionally reduced plaque nitrotyrosine levels. Untreated renal failure mice had increased mineral apposition and bone formation rates, which were reduced by sevelamer but not lanthanum. The study authors concluded that ‘The beneficial effects of La carbonate and sevelamer-HCl on the progression of VC and atherosclerosis in CRF mice could be mainly due to a decrease in phosphate retention and likewise a reduction of arterial Type I collagen expression. The effect of La carbonate differed from that of sevelamer-HCl in that it did not appear to exert its vascular effects via changes in oxidative stress or bone remodeling in the present model’ [Nikolov et al. 2012].
In addition to binding phosphate, sevelamer binds bile acids in vitro and in the gut [Braunlin et al. 2002] [Bays et al. 2008], thus lowering serum LDL cholesterol in people with and without CKD [Braunlin et al. 2002]. These effects of sevelamer are not surprising in view of sevelamer’s similarity to colesevelam, another nonabsorbed resin drug, which was developed as a bile acid sequestrant and hypolipidemic [Bays et al. 2008]. Sevelamer has lowered LDL and total cholesterol in multiple studies of patients with CKD [Gulati et al. 2010] and ESRD [Bleyer et al. 1999; Brandenburg et al. 2010; Braun et al. 2004; Lin et al. 2010; Shantouf et al. 2008; Takenaka and Suzuki, 2005]. High-density lipoprotein (HDL) cholesterol is generally unaffected [Takenaka and Suzuki, 2005]; effects on triglycerides vary among studies. LDL-cholesterol reductions with sevelamer have tended to range from 10% to 20% in predialysis CKD [Di Iorio et al. 2012; Russo et al. 2007; Yilmaz et al. 2012], roughly 20% in incident dialysis patients [Block et al. 2007], and 20% to >30% in prevalent dialysis patients [Bleyer et al. 1999; Braun et al. 2004; Chertow et al. 2002, 2003; Ferramosca et al. 2005; Ferreira et al. 2008; Hervas et al. 2003; Kakuta et al. 2011; Takei et al. 2008], comparable to the ~25% LDL reduction typical of statin use. Reduction of LDL cholesterol reduces a source of vascular inflammation and an atherogenic substance.
Hyperuricemia
High uric acid concentrations induce endothelial oxidative stress, reduce nitric oxide synthase activity [Hong et al. 2012], and are associated with impaired FMV (endothelial dysfunction) in CKD patients [Yelken et al. 2012] although not in healthy adults [Jalal et al. 2012]. Lowering of serum uric acid with allopurinol ameliorated FMV in CKD patients without any change in oxidative markers [Yelken et al. 2012]. Sevelamer has been shown to adsorb urate ions in vitro [Ohno et al. 2009] and to lower serum uric acid in hemodialysis patients [Garg et al. 2005; Ohno et al. 2009]. In the Ohno and colleagues study, sevelamer lowered uric acid significantly only in patients with baseline serum uric acid >7.0 mg/dl [Ohno et al. 2009]. The Garg and colleagues study [Garg et al. 2005] set an a priori threshold for clinically meaningful serum uric acid reduction at −1.5 mg/dl (20–25% of baseline, depending on gender) and observed that 23% of sevelamer recipients but only 10% of calcium recipients experienced at least this level of uric acid reduction (p = 0.02 for treatment group difference). Patients with baseline serum uric acid >8.0 mg/dl had a mean decrease exceeding −2.5 mg/dl [Garg et al. 2005]. In contrast, in the Brandenburg and colleagues study of hemodialysis patients, sevelamer did not affect serum uric acid [Brandenburg et al. 2010]. Serum uric acid in peritoneal dialysis patients [Evenepoel et al. 2009] was lowered by sevelamer hydrochloride but not by calcium carbonate. Since allopurinol treatment to lower serum uric acid is frequently prescribed to retard progression of CKD [Turner et al. 2012], sevelamer’s effect on serum uric acid also may have potential to reduce endothelial dysfunction and its consequences in CKD/ESRD.
Oxalate
Oxalate excretion in urine is a potential source of pathology in patients who have either CKD without overt nephrolithiasis or histories of nephrolithiasis with normal GFR. Different phosphate binders have been studied for their potential to reduce urinary oxalate. In 20 patients with stage 4 or 5 CKD with no history of kidney stones, either sevelamer or calcium carbonate significantly reduced urinary oxalate in a two-period crossover study (3-week exposure to each binder with 1-week washout between) [Caravaca et al. 2007]. Conversely, in an open-label pilot study of 10 patients with enteric hyperoxaluria, normal GFR, and histories of stone formation [Lieske et al. 2008], sevelamer hydrochloride significantly lowered urinary citrate (23%, p = 0.01) and urinary phosphorus (44%, p = 0.0001) and numerically decreased urinary oxalate (17%, p > 0.05) and the oxalate/creatinine ratio (11%, p > 0.05). Sevelamer decreased brushite supersaturation with borderline statistical significance (p = 0.07). The authors suggested that the observed effects of sevelamer on urinary phosphorus, pH, and brushite supersaturation could be clinically significant in patients with calcium phosphate (as opposed to calcium oxalate) stones, and thus sevelamer merits further investigation in additional patient groups [Lieske et al. 2008].
In preclinical studies, lanthanum carbonate has been observed to bind oxalate in vitro at intestinal pH range and to reduce plasma and urine oxalate and nephrocalcinosis in oxalate-treated rats [Robijn et al. 2012].
Endothelial-mediated effects on vascular calcification
Multiple processes in CKD feed into the development of vascular calcification, which is not a merely passive process of mineral crystallization [Sage et al. 2010]. A study investigating patients with moderate to severe predialysis CKD showed that fetuin-A decreases and endothelial dysfunction develops as GFR declines, and concluded that endogenous calcification inhibitors such as fetuin-A mediate a mechanistic relationship between endothelial dysfunction and intimal vascular calcification [Cottone et al. 2010]. Fetuin-A deficiency contributes to atherosclerotic intimal calcification in patients with CKD, atherosclerosis, hyperphosphatemia, and hypercalcemia [Westenfeld et al. 2009]. Another calcification inhibitor, matrix GLa protein, is thought to protect against the medial calcification that is more specific to CKD-MBD-affected vessels. GLa was upregulated in partial-nephrectomy ApoE-/- mice fed a high-phosphate diet [Westenfeld et al. 2009], which may represent a response for acute prevention of medial calcification.
Effects on bone mineral density
Bone loss in CKD-MBD may be linked to development of vascular calcification [Brandenburg et al. 2009]. Improved bone formation rates, bone mineral density (BMD), and trabecular architecture have been reported with sevelamer use in dialysis patients in several studies [Asmus et al. 2005; Ferreira et al. 2008; Lin et al. 2010; Mathew et al. 2007; Raggi et al. 2005]. Simultaneous evaluation of bone quality and vascular calcification is relatively infrequent in binder studies. A study by Asmus and colleagues [Asmus et al. 2005] concurrently examined coronary artery and aortic calcification and vertebral BMD by quantitative computed tomography in 72 adults on hemodialysis randomized to 2 years of calcium carbonate or sevelamer. Calcium recipients had significantly greater increases in coronary artery and aortic calcification than sevelamer recipients. Trabecular BMD significantly decreased in calcium recipients and significantly increased in sevelamer recipients; cortical bone density did not differ between treatments. In a pilot randomized trial in dialysis patients, lanthanum and calcium carbonate did not differ in effects on post-treatment lumbar spine BMD [Toussaint et al. 2011].
Sevelamer in predialysis CKD: primary and pleiotropic effects versus mortality and progression
A recent exploratory study in predialysis CKD patients [Di Iorio et al. 2012] evaluated the effects of sevelamer versus calcium-based binders on mortality or dialysis initiation, vascular calcification, and pleiotropic effects. INDEPENDENT-CKD [Di Iorio et al. 2012] evaluated mortality, progression to dialysis, coronary artery calcification scores, serum albumin, blood lipids, and CRP in 212 consecutive CKD stage 3–4 outpatients randomized to receive sevelamer or calcium carbonate and followed for ≥36 months. The primary endpoint, all-cause mortality, was significantly lower in sevelamer than calcium carbonate recipients, as was a composite endpoint of mortality or dialysis inception. Dialysis inception alone was lower with sevelamer in unadjusted or baseline-covariate-adjusted models, but lost significance in models adjusted for baseline and time-varying covariates. Among patients with non-zero baseline coronary artery calcification (CAC) scores, 24 sevelamer recipients and 2 calcium recipients experienced regression of calcification. New-onset CAC occurred in 5 sevelamer recipients and 45 calcium recipients. Sevelamer but not calcium significantly reduced total and LDL cholesterol. Sevelamer significantly reduced CRP; calcium increased it. The INDEPENDENT-CKD authors conclude that ‘These results show that sevelamer compared to a calcium-containing phosphate binder improves survival in a cohort of incident hemodialysis patients. However, the better outcomes in the sevelamer group may be due to better phosphate control rather than reduction in calcium load.’ [Di Iorio et al. 2012]. INDEPENDENT-CKD is the first study demonstrating a survival benefit for sevelamer use in predialysis CKD patients (albeit this is off-label in the United States and indicated in hyperphosphatemic CKD in many other countries), complementing RIND and INDEPENDENT-HD evidence for this effect in dialysis recipients.
Thus, the pleiotropic effects of sevelamer complement its primary efficacy as a phosphate binder to address multiple aspects of cardiovascular pathophysiology in patients with CKD or ESRD and their antecedent comorbidities of diabetes and/or hypertension.
Ongoing and planned studies (Table 3) continue to explore the pleiotropic effects of sevelamer carbonate or hydrochloride in CKD/ESRD with the following broad foci:
advanced imaging of atherosclerotic plaque inflammation;
effects of sevelamer versus calcium acetate on arterial stiffness and calcification in hemodialysis patients;
FGF-23 and/or PTH in predialysis CKD;
vascular health parameters in predialysis CKD;
anti-endotoxemia and anti-inflammatory effects in patients with HIV infection without CKD.
Recruiting, ongoing, recently completed (unpublished), and planned studies of sevelamer pleiotropic effects from the US ClinicalTrials.gov database.
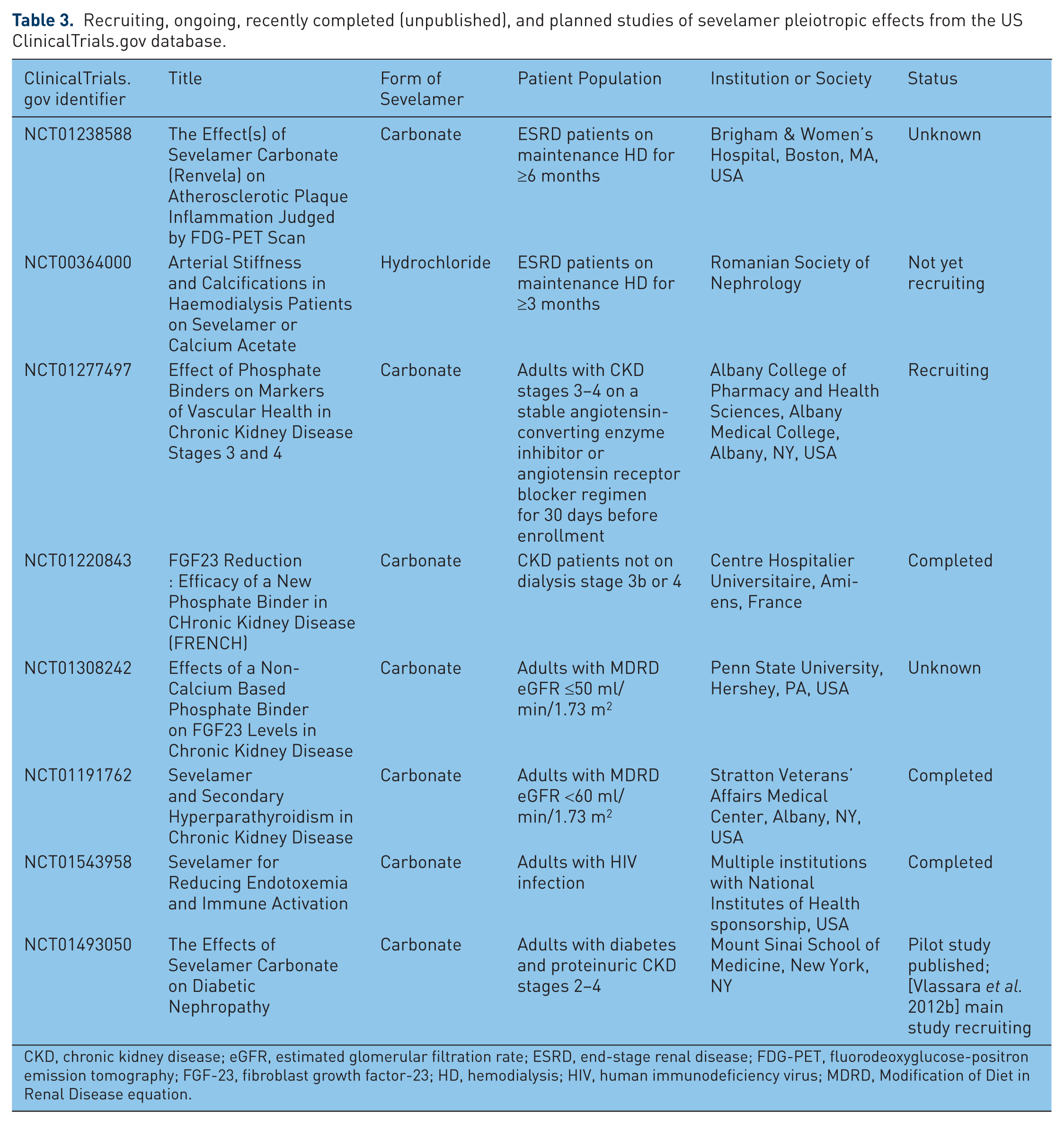
CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; FDG-PET, fluorodeoxyglucose-positron emission tomography; FGF-23, fibroblast growth factor-23; HD, hemodialysis; HIV, human immunodeficiency virus; MDRD, Modification of Diet in Renal Disease equation.
Thus, sevelamer’s ability to lower serum phosphate (with evidence for lowered mortality risk) in ESRD patients (per US labeling) and hyperphosphatemic CKD patients (approved in many non-US countries) is being augmented by current and planned exploratory studies on phosphate-independent effects within and beyond the CKD and ESRD population.
Conclusions
Beyond its labeled effect of serum phosphate reduction in US dialysis recipients and non-US hyperphosphatemic CKD patients of stages 3–5, exploratory data suggest that sevelamer may have additional effects on factors affecting vascular endothelium, such as: arrest of progression of vascular calcification; lowering of total and LDL cholesterol; reduction of FGF-23 (whose elevation is associated with left ventricular hypertrophy); reduction of circulating inflammatory and oxidative molecules, uric acid, and uremic toxins; and reduced blood absorption of AGEs and endotoxin from the gut. It is reasonable to propose that endothelial pleiotropic effects of sevelamer may have contributed to the reduced mortality observed in the RIND and INDEPENDENT studies.
Footnotes
Acknowledgements
The author exerted scientific control throughout manuscript development, provided multiple rounds of substantive content revisions, approved the final published version, and acknowledges the writing assistance of Kim Coleman Healy, PhD, CMPP, of Envision Scientific Solutions, which was contracted by Sanofi (formerly Genzyme) for publication support services. The author received no compensation for manuscript development apart from the provision of publication support services. The manuscript mentions the Sanofi (formerly Genzyme) products sevelamer hydrochloride and sevelamer carbonate.
Funding
The author received no compensation for manuscript development apart from Sanofi provision of publication support services.
Conflict of interest statement
The author discloses speakers’ bureau membership for Cubist, Questcor, Sanofi (formerly Genzyme), and ViiV Healthcare Systems, and advisory board membership for Vifor.