
Abstract
Hereditary bronchiectasis comprises a group of rare monogenic disorders, with cystic fibrosis (CF) and primary ciliary dyskinesia (PCD) representing the major subtypes. Exome sequencing (ES) remains a central modality for molecular diagnosis; however, it leaves more than half of clinically suspected cases unresolved, largely because it cannot reliably detect copy number variations, deep intronic variants, pseudogene-associated variants, and frequent identification of variants of uncertain significance (VUS). This case series describes five hereditary bronchiectasis cases with initial ES-negative results or VUS findings, illustrating the diagnostic utility of targeted genetic approaches. Systematic re-evaluation—including updated bioinformatic pipelines, familial segregation analyses, genome sequencing, RNA sequencing, and functional assays—such as minigene analysis—enabled the reclassification of VUS and the identification of pathogenic variants, leading to definitive diagnoses of PCD or CF in all individuals. Our findings demonstrate that a multimodal strategy integrating ES reanalysis, advanced genomic technologies, and functional validation is critical for resolving previously undiagnosed cases. Furthermore, emerging multiomics integration, artificial intelligence-driven variant interpretation, and global data-sharing frameworks are positioned to further increase diagnostic precision and support the development of targeted therapies for hereditary bronchiectasis.
Keywords
Introduction
Hereditary bronchiectasis encompasses a diverse spectrum of monogenic disorders characterized by bronchial dilation, primarily including cystic fibrosis (CF), primary ciliary dyskinesia (PCD), alpha-1 antitrypsin deficiency, and primary immunodeficiency diseases. 1 These conditions typically manifest in early childhood, with recurrent respiratory infections and bronchiectasis as the predominant clinical features. CF is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. 2 In contrast, PCD exhibits greater genetic heterogeneity, with at least 55 known causative genes identified to date. 3 These genes may follow autosomal recessive, autosomal dominant, or X-linked recessive inheritance patterns. 4 The identification of pathogenic variants through genetic testing is essential for establishing a definitive diagnosis, enabling the development of targeted therapies within precision medicine, and informing genetic counseling.
Exome sequencing (ES), which targets exonic regions and their flanking ±20 bp critical sequences (covering > 98% of coding regions and splice regulatory elements), represents a cost-effective and high-throughput diagnostic approach. It has therefore been widely adopted for the molecular diagnosis of monogenic disorders, including hereditary bronchiectasis. 5 The diagnostic yield of ES varies with phenotype and clinical context, with reported rates ranging from 25% to 40%.5–7 This is consistent with findings from a prospective cohort study involving 87 clinically suspected hereditary bronchiectasis cases, in which ES achieved a diagnostic rate of 41.4% (36/87). 8 However, a substantial diagnostic gap remains: more than 50% of patients with clinically confirmed hereditary bronchiectasis remain genetically undiagnosed after ES, a limitation that impedes timely intervention. 9 This gap is exacerbated by the high frequency of variants of uncertain significance (VUS), which affects nearly 41% of individuals undergoing genetic testing; such inconclusive findings create clinical ambiguity, delaying treatment decisions and complicating genetic counseling. 10 Compounding this challenge, genetic diagnosis of CF and PCD, the principal subtypes of hereditary bronchiectasis, is uniquely challenged by disease-specific complexities, including extensive locus heterogeneity, variable penetrance, and limited functional characterization of rare alleles, all of which increase the VUS burden in these conditions. 10
Emerging evidence suggests that reanalysis of ES data—integrating updated bioinformatics pipelines, recent literature, and refined phenotype–genotype correlations—can increase diagnostic yield by 15%. 11 However, most studies on ES reanalysis have focused on pediatric and neurological disorders, and relatively few have examined post-ES diagnostic strategies in respiratory genetic diseases. In the following cases of hereditary bronchiectasis from Peking Union Medical College Hospital, we present the clinical decision-making processes and subsequent genetic diagnostic strategies used for patients with negative or VUS results from ES.
The reporting of this study conforms to the CARE guidelines (Supplemental Material). 12
Case 1: VUS reclassification
A 37-year-old male presented with a recurrent productive cough. He had been diagnosed with situs inversus in early childhood. Chest computed tomography (CT) revealed bronchiectasis predominantly involving the right middle and lower lobes, while paranasal sinus CT demonstrated chronic maxillary sinusitis. His clinical presentation was consistent with Kartagener syndrome. Nasal nitric oxide (nNO) measurement showed normal values (314.8 nL/min). ES identified compound heterozygous variants in ODAD4 (NM_031421.5):c.246+1G>T, which was classified as likely pathogenic (LP) according to the American College of Medical Genetics and Genomics (ACMG) guidelines, supported by PVS1 (predicted loss-of-function in a gene where loss-of-function is a known disease mechanism) and PM2 (extremely low frequency in the ESP, 1000 genomes, and ExAC databases). 13 The second variant, ODAD4 (NM_031421.5):c.1528+1G>T, was classified as a VUS based on PVS1 and BS1 (East Asian allele frequency in gnomAD: 0.006858). 14
Subsequent Sanger sequencing of the patient’s sister demonstrated that she carried variant 1 but not variant 2, thereby establishing their trans configuration (PM3). The patient’s Kartagener syndrome phenotype provided additional supporting evidence for a PCD diagnosis (PP4). On this basis, variant 2 was reclassified as LP with the following supporting criteria: PVS1 + BS1 + PM3 + PP4. Given that ODAD4 is an established PCD-causative gene and the genetic findings were consistent with the patient’s clinical manifestations, the patient received a definitive diagnosis of PCD. 15
This case illustrates that when a phenotype-associated variant is initially reported as a VUS, systematic re-evaluation, including family segregation studies and refined phenotype–genotype correlation, may provide sufficient evidence for reclassification.
Case 2: Limitations of ES in the detection of copy number variations
A 19-year-old male presented with a history of recurrent pulmonary infections since early childhood. Chest CT revealed diffuse bronchiectasis predominantly involving the lingula and left lower lobe, accompanied by dextrocardia and situs inversus of the liver and spleen (Figure 1). At 18 years of age, he underwent functional endoscopic sinus surgery for refractory chronic sinusitis. Semen analysis showed severe oligoasthenozoospermia, with only one immotile sperm observed per high-power field. His clinical findings were consistent with Kartagener syndrome. ES identified a single heterozygous variant in DNAH5 (NM_001369.3):c.7769G>A (p.Gly2590Asp), classified as a VUS according to ACMG criteria, supported by the following evidence: PM2 + PP3 (multiple computational predictions consistently supporting a deleterious effect on protein function) + PP4. Sanger sequencing of both parents confirmed maternal inheritance of this variant.
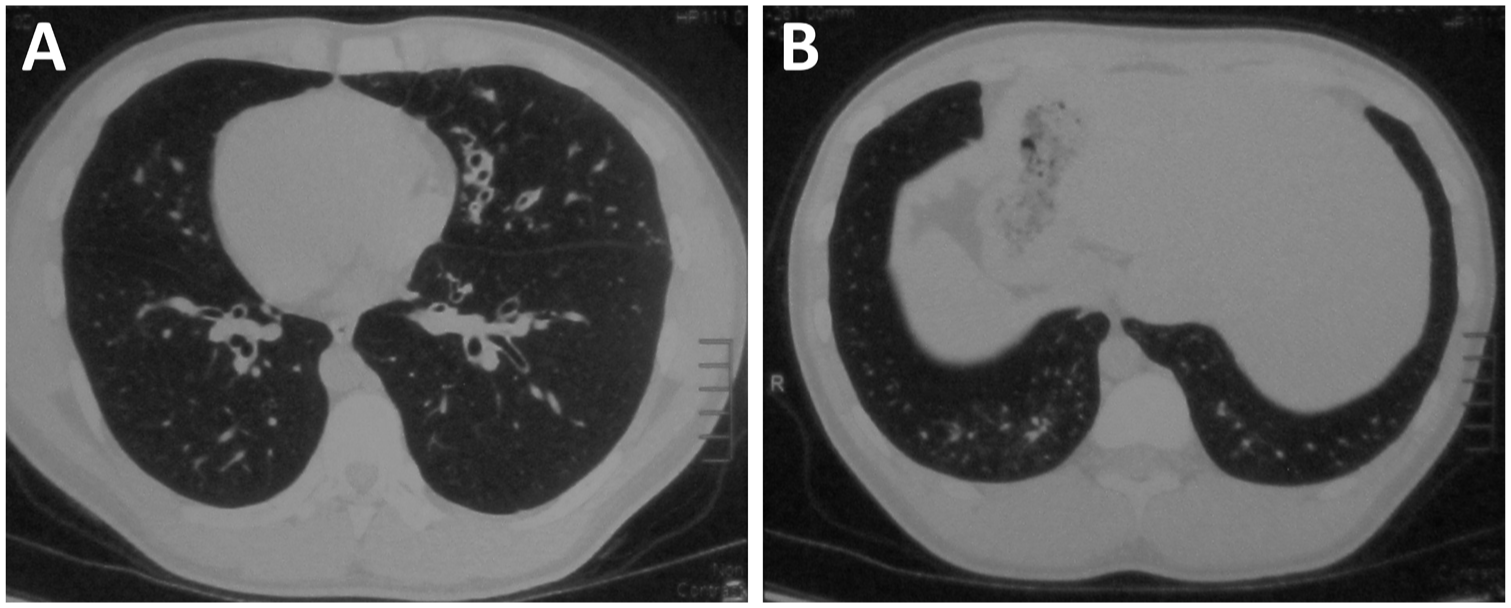
Chest CT of Case 2. (a) Chest CT demonstrates diffuse bronchiectasis in both lungs, predominantly in the lingula and left lower lobe. (a and b) Dextrocardia and situs inversus of the liver and spleen are observed.
Given the limited sensitivity of ES for detecting copy number variations (CNVs), genome sequencing (GS) was performed to identify a potential second pathogenic variant in DNAH5. This analysis revealed a paternally inherited intragenic duplication spanning exons 42–44 (verified by quantitative polymerase chain reaction (qPCR) of genomic DNA), introducing a 566-nucleotide frameshift that disrupts the open reading frame. According to the ACMG guidelines, this CNV was classified as pathogenic. With the addition of PM3 evidence, the DNAH5 (NM_001369.3):c.7769G>A variant was reclassified as LP. Given that DNAH5 is an established PCD-causative gene and the genetic results were concordant with the patient’s clinical phenotype, the patient received a definitive diagnosis of PCD. 16
This case underscores that ES has significant limitations in detecting clinically relevant structural variants (SVs), including CNVs, chromosomal inversions, translocations, and short tandem repeats. For patients with strong clinical suspicion of hereditary bronchiectasis but negative or inconclusive ES results, GS should be considered as a subsequent diagnostic step due to its superior ability to identify such complex genomic alterations.
Case 3: Limitations of ES in deep intronic variants detection
A 29-year-old male presented with recurrent productive cough since the neonatal period (Figure 2(a)). He had noted salt deposits on his skin after sweating since childhood. Chest high-resolution computed tomography (HRCT) revealed diffuse bronchiectasis in both lungs, predominantly in the upper lobes, with mucus plugs occupying the dilated airways (Figure 2(b)). He also had a 6-year history of chronic sinusitis (Figure 2(c)). He was born prematurely at gestation period of 28 weeks and required prolonged hospitalization until 3 years of age. He was conceived naturally and has one healthy son. His parents are nonconsanguineous. Among his four siblings, a 23-year-old sister was found to have bilateral bronchiectasis (predominantly in the left upper lobe) on HRCT during evaluation for chronic cough (Figure 2(d)), although she denied symptoms of sinusitis or otitis media (Figure 2(a)). Sweat chloride testing showed elevated bilateral results (70 mmol/L and 84 mmol/L, respectively). However, ES performed in 2019 did not detect any CFTR-related variants.

Family pedigree and radiological findings of Case 3. (a) Family pedigree showing an autosomal recessive inheritance pattern: the affected proband (II-3, arrow) and sister (II-4) exhibit a CF phenotype, whereas other siblings remain unaffected. (b) Chest HRCT of the proband (II-3) reveals severe diffuse bronchiectasis with upper lobe predominance and associated mucoid impaction. (c) Paranasal sinus CT of the proband (II-3) demonstrates characteristic pansinusitis. (d) Chest HRCT of the affected sister (II-4) shows bilateral multifocal bronchiectasis, with the most severe involvement in the left upper lobe.
The patient’s elevated sweat chloride levels strongly supported a clinical diagnosis of CF, despite the negative initial ES results. In August 2023, trio-based family ES was repeated at another commercial testing company, which identified a homozygous CFTR (NM_000492.3):c.3718-2477C>T variant inherited from both parents. According to the ACMG guidelines, this variant was classified as LP, with supporting evidence including PS3 (well-established functional studies demonstrating damaging effects) + PM2 + PM3 + PP1 (co-segregation with the disease in the family). Genetic testing further confirmed the CF diagnosis. 17 Subsequent validation demonstrated that the patient’s sister carried the same homozygous CFTR variant and was likewise diagnosed with CF.
This case underscores that ES has inherent limitations in detecting deep intronic variants because probe coverage of noncoding regions is incomplete. It also highlights that the interpretation and classification of pathogenic variants continue to evolve as additional functional and clinical evidence emerges. For patients with a high clinical suspicion of hereditary bronchiectasis and initially negative ES results, periodic repeat ES (with updated probe sets), or reanalysis of existing ES data—particularly when clinical suspicion remains strong—are essential for securing an accurate genetic diagnosis.
Case 4: Limitations of ES in distinguishing variants in functional genes versus pseudogenes
A 20-year-old male presented with recurrent productive cough since 4 years of age. Bronchiectasis was first identified on chest CT at 12 years of age. He reported a 10-year history of chronic sinusitis but denied neonatal pneumonia or nasal polyposis. nNO measurement showed markedly reduced levels (10.5 nL/min). Semen analysis revealed a complete absence of progressively motile sperm (0% for grade A + B sperm; normal reference ⩾30%), consistent with asthenozoospermia. HRCT demonstrated diffuse bronchiectasis predominantly involving the lower lobes (Figure 3). ES did not identify any pathogenic or likely pathogenic variants explaining the patient’s phenotype.

Chest HRCT of Case 4. Chest HRCT (a and b) demonstrates diffuse bronchiectasis predominantly involving the lower lobes (b).
Although the patient was clinically suspected to have PCD, initial ES did not detect any PCD-associated pathogenic variants. Subsequent GS revealed compound heterozygous variants in HYDIN (NM_001270974.1):c.6685C>T (p.Arg2229*), inherited paternally (ACMG classification: Pathogenic [PVS1 + PM2 + PP4]); and c.1670+2T>A, inherited maternally (ACMG classification: Pathogenic [PVS1 + PM2 + PP4]). HYDIN is a known PCD-causative gene; however, its high sequence homology with the pseudogene HYDIN2 introduces significant diagnostic challenges, as next-generation sequencing may yield false-positive or false-negative results owing to cross-hybridization or misalignment. 18 Confirmatory testing—such as targeted Sanger sequencing, long-read sequencing, RNA sequencing (RNA-seq), or ancillary techniques (e.g., immunofluorescence for SPEF2 protein)—is often necessary to establish a definitive diagnosis.19,20 In this patient, GS followed by Sanger validation, combined with concordant clinical phenotypic and genetic evidence, confirmed the diagnosis of PCD.
This case underscores the diagnostic limitations of ES in differentiating variants located in functional genes from those in highly homologous pseudogenes. To optimize sequencing strategies, it is critical to pre-emptively identify whether key diagnostic genes of interest have homologous genes or high-similarity sequences that may interfere with mapping and variant calling, and to incorporate appropriate confirmatory assays when such genes are implicated.
Case 5: Functional assays reclassify VUS as pathogenic
A 32-year-old male presented with recurrent wet cough since childhood. His medical history included chronic sinusitis and nasal polyps beginning in early childhood, although he had no history of neonatal pneumonia or otitis media. He was conceived naturally and has a healthy daughter. nNO levels were markedly reduced (9.2 nL/min). Chest HRCT demonstrated bilateral bronchiectasis, most prominent in the middle and lower lobes, along with atelectasis of the right middle lobe (Figure 4(a) and (b)). Paranasal sinus CT revealed inflammatory changes involving multiple sinuses (Figure 4(c)). Trio-based ES detected compound heterozygous variants in RSPH4A (NM_001010892.2):c.1468C>T (p.Arg490*), inherited paternally and classified as pathogenic (PVS1 + PM2 + PM3); and c.1663-3C>G, inherited maternally and classified as a VUS (PM2 + PM3 + PP3).

Radiological findings and minigene assay results of Case 5. (a and b) Chest HRCT demonstrates bilateral bronchiectasis with predominant middle and lower lobe involvement, accompanied by right middle lobe atelectasis. (c) Paranasal sinus CT reveals pansinusitis with inflammatory changes. (d) Schematic representation of the minigene construct, illustrating the genomic fragment (variant site indicated by red arrow) inserted into the pCAS2 vector between exons A and B. (e) Agarose gel electrophoresis shows aberrant splicing in the mutant, yielding a truncated transcript (~936 bp) versus wild-type (~1068 bp). (f) Sanger sequencing confirms two distinct abnormal transcripts in the mutant. (g) Splicing pattern analysis demonstrates the following: (i) Partial exon 3 skipping with complete exon 4 exclusion, and (ii) Complete exon 4 skipping (direct exon3–exon5 joining). Pathogenic variant site is highlighted in yellow.
The patient’s clinical history strongly supported PCD diagnosis; however, the genetic diagnosis remained inconclusive because only one pathogenic variant was identified alongside a VUS. RSPH4A is a known PCD-associated gene, and the c.1663-3C>G variant—located 3 bp upstream of the exon 4 splice acceptor site—was predicted to significantly alter splicing (SpliceAI score 0.81; cutoff ⩾0.5). 21 To validate its functional impact, wild-type and mutant minigene plasmids were engineered using the pCAS2 vector, incorporating exon 3 through exon 5 with flanking intronic regions (Figure 4(d)). Following transfection into HEK293 cells, reverse transcription-polymerase chain reaction (RT-PCR) analysis revealed a shorter transcript in the mutant compared with the wild type (Figure 4(e)). Sanger sequencing revealed two aberrant splice isoforms (Figure 4(f)): one isoform utilized a cryptic donor within exon 3 paired with an acceptor at the exon 5 start site, whereas the second isoform exhibited complete skipping of exon 4 (Figure 4(g)). These findings confirmed that c.1663-3C>G disrupts normal mRNA processing by causing partial truncation of exon 3 and complete skipping of exon 4. With functional evidence of aberrant splicing (PVS1) and a clinical phenotype highly consistent with PCD (PP4), the c.1663-3C>G variant was reclassified as pathogenic (PVS1 + PM2 + PM3 + PP3 + PP4) according to ACMG guidelines, thereby establishing a definitive genetic diagnosis of PCD.
This case demonstrates that functional assays play a critical role in resolving the pathogenicity of VUS identified by ES, enabling conclusive diagnoses in clinically affected patients with otherwise inconclusive genetic findings.
Discussion
These cases highlight the downstream diagnostic strategies for patients with hereditary bronchiectasis who initially receive negative ES results (Table 1). For such patients with suspected hereditary bronchiectasis and negative ES findings, systematic clinical re-evaluation and comprehensive family history assessment constitute the foundation of diagnostic reasoning. Phenotypic characterization should include detailed evaluation of medical history (including age at symptom onset, frequency and sites of infections, and associated conditions such as neonatal pneumonia, subfertility, chronic sinusitis, or otitis media) and family history (consanguinity and presence of similarly affected relatives), coupled with targeted physical examinations (e.g., assessment for situs inversus). 1 Abnormal results from key ancillary tests can substantially enhance the clinical prediction of specific genetic disorders and can help guide subsequent genetic investigations. For example, a sweat chloride concentration >60 mmol/L supports a diagnosis of CF; characteristic ciliary ultrastructural defects on transmission electron microscopy confirm PCD; low serum alpha-1 antitrypsin levels indicate alpha-1 antitrypsin deficiency; and reduced lymphocyte counts and immunoglobulin levels suggest immunodeficiency.1,22,23
Clinical phenotypes and subsequent genetic diagnostic strategies for the five studied cases.

CF, cystic fibrosis; CNVs, copy number variations; ES, exome sequencing; GS, genome sequencing; LP, likely pathogenic; nNO, nasal nitric oxide; path, pathogenic; PCD, primary ciliary dyskinesia; VUS, variant of uncertain significance.
Negative ES results in patients with hereditary bronchiectasis may be recorded in two major scenarios: (1) failure to identify candidate pathogenic genes or variants, which can be further subclassified as either (i) detection of only a single pathogenic variant in autosomal recessive conditions or (ii) complete absence of candidate pathogenic genes or variants; and (2) identification of VUS. Diagnostic approaches should be tailored to these specific circumstances.
For patients with hereditary bronchiectasis in whom no candidate pathogenic genes or variants were identified by initial ES, at least one re-analysis of existing ES data is recommended—particularly if the initial analysis was performed more than 12 months earlier. According to ACMG guidelines, ES re-analysis may be initiated through three mechanisms: (1) clinician-initiated re-evaluation prompted by patient care needs; (2) research-driven analyses conducted by scientific teams; or (3) periodic laboratory-led reviews. 24 Positive findings from re-analysis are primarily recorded in two settings: (i) variants previously undetected due to technical limitations (such as indels, noncoding variants in exon-flanking regions, or CNVs), and (ii) newly established gene–disease relationships supported by evolving evidence from multidisciplinary re-evaluation of emerging evidence (e.g., advances in gene–disease relationship studies or segregation analysis data). 25 Case 3 of our series exemplifies the value of repeat ES testing performed 4 years after the initial test. Due to continuous improvements in CFTR variant detection and expanded ES probe coverage, the repeat ES identified a pathogenic variant that was undetected in the initial analysis, enabling a definitive genetic diagnosis. 17 This emphasizes the clinical importance of periodic ES data re-analysis or repeat testing, especially given the rapid evolution of genomic technologies and variant interpretation frameworks.
If ES re-analysis remains negative, alternative genetic testing technologies may be considered for further evaluation. Relevant detection techniques include the following:
Short-read genome sequencing
Short-read genome sequencing (srGS) provides comprehensive coverage of the entire genome, enabling simultaneous detection of coding and noncoding variants as well as SVs, including CNVs, inversions, and short tandem repeats. 26 Multiple studies have shown that srGS can increase the diagnostic yield by approximately 10% in ES-undiagnosed cases.27,28 Despite technical limitations—such as challenges in interpreting the pathogenicity of noncoding variants and SVs, higher costs, and complex data analysis—its ability to detect multiple variant types in a single assay enhances its cost-effectiveness as sequencing technology advances and costs decline. Case 2 in our series illustrates the limitations of ES in detecting CNVs; for patients with a typical phenotype, srGS may therefore be considered to facilitate diagnosis.
Long-read genome sequencing
Long-read genome sequencing (lrGS) generates single-molecule DNA reads ranging from ~1 kb to several Mbs in length. Compared with short-read sequencing, this technology more accurately maps repetitive genomic regions, thereby enabling robust detection of pathogenic structural variants. Additionally, it supports haplotype analysis of variants without requiring parental samples. 29 lrGS can improve diagnostic rates in ES-negative cases; however, its clinical application remains limited by higher costs, complex data-analysis workflows, and greater challenges in clinical interpretation. 25 Case 4 in our series highlights the limitations of ES probes in distinguishing functional genes from pseudogenes, underscoring that third-generation sequencing (e.g., lrGS) can assist in diagnosing variants in hotspot genes affected by pseudogene interference, such as HYDIN and NCF1.
RNA sequencing
RNA-seq serves as an important complementary technique to DNA sequencing, offering unique diagnostic value. This method does not rely on prior knowledge of DNA variants and can directly identify pathogenic genes by detecting aberrant expression or splicing abnormalities. When integrated with DNA sequencing data, RNA-seq can help elucidate the functional impact of splicing and noncoding variants, uncover missed second pathogenic alleles in recessive disorders, and aid the discovery of novel disease-causing genes in undiagnosed cases. 30 However, its application is constrained by tissue-specific expression: for example, in patients with PCD, many cilia-related genes show low or absent expression in peripheral blood, necessitating bronchial or nasal mucosal samples for RNA-seq analysis. 31
For patients with VUS, the following workflow is recommended to strengthen evidence of pathogenicity: First, re-analyze ES data by integrating the latest databases and guidelines, including but not limited to enhancing pathogenicity evidence for variants and genotype-driven data reanalysis. In case 1 of this series, one pathogenic ODAD4 variant and one VUS were identified. Familial segregation analysis confirmed that the two variants were in trans, providing PM3 evidence for the VUS and enabling a definitive genetic diagnosis.
Subsequently, RNA-seq analysis should be performed to evaluate the impact of the variant on gene expression or splicing. When necessary, functional analyses can be conducted, including (1) construction of eukaryotic expression vectors to assess protein expression levels 32 ; (2) minigene assays, in which partial exons and introns containing the mutant site are cloned into a vector, transfected into cell lines, and analyzed via RT-qPCR or sequencing to detect aberrant splicing 33 ; and (3) generation of gene-knockout animal models for phenotypic assessment and investigation of pathogenic mechanisms. 34 In case 5, a minigene assay confirmed aberrant splicing (PS3), enabling reclassification from VUS to LP and securing a definitive diagnosis.
Advances in genetic-testing technologies have provided new diagnostic pathways for ES-negative patients with suspected hereditary bronchiectasis (Figure 5). Current evidence supports a stepwise approach for clinically suspicious cases with negative ES results: first, re-analysis of ES data; second, implementation of srGS combined with functional analysis or RNA-seq as subsequent testing strategies; and third, consideration of lrGS when SVs are suspected. In clinical practice, we recommend tailoring the testing approach to the patient’s phenotype via multidisciplinary discussion involving medical geneticists, with the goal of developing a personalized diagnostic plan.

Diagnostic algorithm for suspected hereditary bronchiectasis with negative ES results.
Perspective
The diagnostic strategies outlined above have demonstrated efficacy in resolving complex cases of hereditary bronchiectasis, yet they also reflect broader developments and future directions in clinical genetics aimed at addressing the central challenge of VUS interpretation. With ongoing technological advances and strengthened global collaborative efforts, we anticipate transformative progress in the interpretation and clinical management of VUS, which will further enhance diagnostic precision in hereditary bronchiectasis and other monogenic disorders.
Technological innovation: From single-modality testing to multiomics integration
Future genetic testing strategies for hereditary bronchiectasis will increasingly shift toward multiomics integration. Sequential or parallel analyses of DNA, RNA, methylation, metabolomics, and proteomics can overcome the inherent limitations of conventional single-modality genetic testing. Integrated multiomics analysis enables systematic correlation of multidimensional data to circumvent these constraints. This integration is not merely data aggregation but rather employs bioinformatic frameworks to cross-validate genomic variants with dynamic transcriptomic, epigenomic, proteomic, and metabolomic signatures. For example, Murdock et al. achieved an overall diagnostic yield of 17% in 115 patients through transcriptome-guided analysis, successfully identifying pathogenic variants that were undetectable by conventional methods—such as deep intronic variants in PQBP1 and a 307-kb deletion in KANSL1. 35 Similarly, Alaimo et al. applied integrated metabolomic-genomic analysis to reinterpret variants in 170 patients, reclassifying 24 variants and confirming diagnoses in 21 cases (diagnostic yield ~12.3%). 36
For hereditary bronchiectasis specifically, multiomics integration may, for instance, connect CFTR deep intronic variants (identified through DNA sequencing) with aberrant splicing patterns (validated via RNA-seq) and impaired chloride transport (quantified through proteomic or functional assays), collectively providing a more comprehensive evidence base for variant classification and treatment-response prediction.
Large-scale application of functional genomics technologies will also become pivotal for addressing VUS. Multiplexed assays of variant effects enable high-throughput functional evaluation of all possible single-nucleotide variants, generating comprehensive variant-effect maps that provide systematic functional annotations for bronchiectasis-associated genes (e.g., DNAH5). 37 Concurrently, organoid-based functional validation models (e.g., airway epithelial organoids) can simulate the physiological impact of variants, particularly for assessing the pathogenicity of variants related to ciliary structure or function.
Deep integration of computational modeling and artificial intelligence
Advances in machine-learning algorithms are reshaping the paradigms of variant interpretation. Deep-learning models (e.g., AlphaMissense and REVEL) trained on large-scale genome–phenotype datasets have markedly improved the accuracy of missense variant prediction, with some models achieving up to 89% accuracy. 37 Future efforts will benefit from developing disease-specific prediction models for hereditary bronchiectasis, incorporating airway epithelial gene-expression profiles, protein–protein interaction networks, and detailed clinical phenotypes to construct a multidimensional “gene–variant–phenotype” predictive framework.
Bayesian approaches also show considerable promise in variant classification. By integrating multisource evidence—including population frequencies, functional predictions, and clinical data—probabilistic models can support dynamic VUS reclassification. 38 For example, in neurofibromatosis type 1 testing, a clinical variant model based on Bayesian methodology incorporated phenotypic data from 333 VUS cases, population allele frequencies, and functional prediction results. Hierarchical Bayesian inference enabled estimation of gene penetrance and phenotype simulation rates, ultimately reducing VUS reporting by 49% and affecting 12,094 patients. Within this framework, dual-level patient and variant scoring reclassified VUS as benign or pathogenic, with 95.9% categorized as benign, substantially reducing clinical uncertainty. 39
Data sharing and international collaboration
Global genomic data-sharing networks represent a critical mechanism to address VUS interpretation challenges. 40 Although databases such as ClinVar and gnomAD currently contain over 3 million variant annotations, disease-specific data for hereditary bronchiectasis remain limited. There is an urgent need to establish dedicated databases for respiratory genetic disorders that integrate detailed clinical phenotypes, functional data, and multiomics datasets to enhance the accuracy of variant interpretation.
Conclusion
In summary, the diagnostic paradigm for addressing negative genetic testing results and VUS in hereditary bronchiectasis is shifting from isolated technological modalities toward multidimensional integration. Through synergistic progress in multi-omics technologies, computational modeling, and global data-sharing frameworks, we anticipate a marked reduction in VUS rates within the next 5–10 years. These advances will support the transition from “variants of uncertain clinical significance” to “precise diagnosis and targeted therapy,” thereby opening new avenues for prevention, diagnosis, and therapeutic management of hereditary bronchiectasis.
Supplemental Material
sj-pdf-1-tar-10.1177_17534666261422041 – Supplemental material for Genetic strategies for negative or variant of uncertain significance findings in exome sequencing in hereditary bronchiectasis: a case series
Supplemental material, sj-pdf-1-tar-10.1177_17534666261422041 for Genetic strategies for negative or variant of uncertain significance findings in exome sequencing in hereditary bronchiectasis: a case series by Wangji Zhou, Yixuan Li, Qiaoling Chen, Yaqi Wang, Aoyue Li, Wanqing Lu, Xiaogang Li, Kai-Feng Xu, Xue Zhang, Yaping Liu and Xinlun Tian in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.