
Abstract
Background:
Pulmonary alveolar microlithiasis (PAM) is a rare autosomal recessive disorder caused by SLC34A2 variants, characterized by diffuse alveolar calcium phosphate deposits. While the SLC34A2 mutation spectrum has been well-documented, the distinct variant landscape in Chinese patients remains unclear.
Objectives:
This study aims to report three newly identified PAM cases and describe the SLC34A2 mutation spectrum of Chinese PAM patients through a systematic review.
Design:
We documented the diagnosis and treatment processes and genetic variations of three PAM cases for reporting. Furthermore, we searched academic websites for published PAM cases with SLC34A2 variants and extracted clinical and genetic data for analysis.
Methods:
We employed whole-exome sequencing to identify genetic mutations of these three patients. We systematically searched PubMed, Web of Science, China National Knowledge Infrastructure, and Cochrane Library for published PAM cases with SLC34A2 mutations. Clinical and genetic data were extracted into an Excel database and analyzed using SPSS 23.0 software (IBM, Armonk, NY, USA).
Results:
Among the three cases we reported, two homozygous mutations in SLC34A2-c.910A>T (p.Lys304*) in exon 8 and c.575C>A (p.Thr192Lys) in exon 6 were identified. Analysis of 27 Chinese and 49 non-Chinese PAM patients revealed similar clinical manifestations, but a strikingly distinct genetic spectrum. Compound heterozygous mutations predominated in Chinese patients, while only two cases of compound heterozygous mutations were found in non-Chinese patients. Deletion/insertion mutations are the most common in non-Chinese patients (19/47, 40.4%), whereas nonsense mutations are the most frequent in Chinese patients (12/20, 60%). Further analysis of the reported SLC34A2 mutation sites in Chinese PAM patients showed hotspot regions in exons 5, 6, and 8, with c.910A>T in exon 8 being a unique gene screening target in Chinese patients.
Conclusion:
This study delineates a distinct spectrum of SLC34A2 mutations in Chinese PAM patients, highlighting the importance of ethnicity-specific genetic screening in PAM diagnosis.
Introduction
Pulmonary alveolar microlithiasis (PAM) is a rare progressive lung disease characterized by the extensive formation of tiny calcium phosphate deposits (microliths) in the alveoli. First described by Malpighi in 1686 1 and named by Puhr in 1933, 2 PAM is an autosomal recessive disorder mainly caused by mutations in the solute carrier family 34 member 2 (SLC34A2) gene.
The SLC34A2 gene, which encodes sodium-dependent phosphate transport protein 2B, plays an important role in actively transporting phosphate into cells via Na+ cotransport 3 and has been identified as the causative gene for PAM.4,5 Pulmonary surfactants are produced by type II alveolar cells, which are mainly composed of phospholipids. Subsequently, phosphate is produced by the degradation of phospholipids and released into the alveolar space. 6 NaPi-IIb is not only expressed in the lungs, but can also be detected in the pancreas, kidneys, small intestine, ovaries, testes, prostate, and breast.3,7 PAM patients may also have extrapulmonary manifestations. Calcification in other sites such as the tricuspid valve, 8 pericardium, 9 gastric mucosa,10,11 kidney, 12 urethra, gallbladder, 13 sympathetic nervous system, testes, 14 and seminal vesicles 15 have been reported.
Most PAM patients are initially asymptomatic at the early stage and are incidentally diagnosed by chest imaging. As the disease progresses, symptoms such as cough, dyspnea, and eventually respiratory failure may occur. Lung imaging of patients with PAM often shows a typical “sandstorm” appearance due to the diffuse distribution of microliths. PAM can be definitively diagnosed based on typical imaging manifestations of high-resolution computed tomography (CT), including “intra-alveolar calcifications, micronodules, ground-glass opacities, thickened septa, subpleural cysts,” combined with SCL34A2 biallelic mutation. In the absence of or with negative results of genetic testing, detection of microliths by bronchoalveolar lavage (BAL) or pathologic biopsy is required to confirm the diagnosis. PAM treatment is primarily supportive, and lung transplantation is the only effective treatment for end-stage disease. 16
The variant spectrum of the SLC34A2 gene in Chinese PAM patients has not been previously reported. Understanding the genetic basis is critical to improve patient outcomes and develop effective therapies. In this paper, we describe the clinical course and SLC34A2 gene mutations of three new patients we recently identified. In addition, through a comprehensive literature review, we analyzed the genetic characteristics of 27 Chinese and 49 non-Chinese PAM patients, comparing their variant spectra and clinical presentations.
Materials and methods
Subjects
Three unrelated patients were enrolled in this study because of characteristic pulmonary imaging findings and varying degrees of dyspnea. Peripheral blood samples were collected from the patients for genetic testing.
Genetic testing
The whole-exome high-throughput sequencing service was provided by the project to improve the diagnosis and treatment capacity of rare diseases, supported by the Central Special Lottery Fund. The data were analyzed by the Verita Trekker® mutation detection system and the Enliven® variant annotation system, independently developed by BerryGenomics (https://www.berrygenomics.com). Suspected dynamic mutations identified by whole-exome sequencing were analyzed by PCR and capillary electrophoresis. According to the American College of Medical Genetics and Genomics (ACMG) guidelines 17 and ClinGen Sequence Variant Interpretation Expert Group recommendations on the application of guideline standards,18–20 we screened HPO (https://hpo.jax.org), OMIM (http://www.omim.org), GHR (https://medlineplus.gov/genetics/), and other public databases to find clinically available phenotypes or disease-related variants. Mutation Taster (http://www.mutationtaster.org/), Poly Phen-2 (http://genetics.bwh.arvard.edu/pph2), and Franklin (https://franklin.genoox.com/clinical-db/home) were used to predict the effects of these variants. SWISS-MODEL (https://swissmodel.expasy.org/) was used to predict the protein structure, and PyMOL (https://www.pymol.org) was used to generate diagrams of the mutant protein.
Literature search strategies
The keywords “Pulmonary alveolar microlithiasis and SLC34A2” were searched in PubMed (https://pubmed.ncbi.nlm.nih.gov), Web of Science (https://www.webofscience.com/), China National Knowledge Infrastructure (https://www.cnki.net), and Cochrane Library (https://www.cochranelibrary.com) to retrieve articles and additional records identified through reference lists from published papers. Researchers in our group reviewed these articles, selected the literature reporting SLC34A2 mutations in PAM patients, and excluded duplicate reports and cases. Finally, we sorted out the genetic information of Chinese and non-Chinese PAM patients.
Diagnosis of PAM
There are currently no consensus or guidelines for the diagnosis of PAM. According to the published review, 16 the diagnosis can be made if the following conditions are met: (1) Pulmonary imaging shows “Sandstorm” appearance or typical changes including alveolar calcification, micronodules, interlobular septal thickening, and subpleural cysts. (2) Genetic testing confirmed a biallelic variant of SCL34A2. If genetic testing is unavailable or the result of genetic testing is negative, the diagnosis can also be confirmed by the presence of alveolar microlithiasis in BAL or lung biopsy.
Data collection and analysis
Data from eligible articles were extracted and summarized in an Excel spreadsheet, including: (1) Article title and publication year. (2) Demographic information of patients: gender, age, and family history. (3) Clinical symptoms: cough, sputum, dyspnea, and so on. (4) Pulmonary function tests (forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO)) and radiologic manifestations. (5) Genetic information: SLC34A2 variants, mutation types, and protein changes. SPSS 23.0 software (IBM SPSS, USA) was used for data analysis. The Kolmogorov–Smirnov test was used to assess the normality of all continuous variables. Continuous variables were expressed as mean ± standard deviation or median and interquartile range (IQR); categorical variables were presented as frequency (percentage). Student’s t-test or the Mann–Whitney U test was used for continuous variables; the Chi-square test (χ2), Yates’s correction, or Fisher’s exact test was used for categorical variables.
Case presentation
Supplemental Table 1 summarizes the demographic, clinical, and genetic characteristics of three patients diagnosed with PAM.
Case 1
This male patient was found to have an abnormal chest radiograph in his twenties, but remained asymptomatic at first. In 2014, at age 47, he was hospitalized for evaluation of a persistent dry cough. His parents were cousins, and his mother had a history of tuberculosis and died from esophageal cancer. He had a 30-pack-year smoking history and was exposed to dust due to his work in construction.
His pulmonary function was normal upon initial admission (FVC 111% predicted, DLCO 97% predicted). CT revealed interstitial calcification in both lungs (Figure 1(a)), and a biopsy confirmed microlith deposition in the interstitial and alveolar spaces. Echocardiography and ultrasound of the liver, gallbladder, pancreas, spleen, kidneys, and ureters were unremarkable.

Chest CT and DNA sequence results for the SLC34A2 gene of three patients with PAM pulmonary alveolar microlithiasis. (a) CT of case 1 in 2014, when first visited our hospital: bilateral pulmonary interstitial thickened and both lung fields calcified significantly, mainly in the middle and lower lobes. (b) CT of case 1 in 2023: similar to 2014, no obvious progress. (c) CT of case 2 in 2006: bilateral lung lesions were mild, and calcification was small. (d) CT of case 2 in 2023: The lesions of both lungs were significantly aggravated, showing a paving stone-like change, and the interlobular septum was thickened. CT of case 3 in 2023 showed that (e) double upper lobe and (f) lower lobe calcified significantly, and diffuse punctate density shadows mainly located in the subpleural. (g)–(i) SLC34A2 variants in three patients are present in homozygous form. Top row: SLC34A2 DNA reference sequence (NM_006424.3). Bottom row: Mutated DNA sequences of three patients. The arrows indicate the locations of the variants.
The patient was diagnosed with alveolar microlithiasis and received symptomatic treatment. Over time, he developed exertional dyspnea, intermittent cough, and sputum production. The re-examination of pulmonary function tests (PFTs) in 2023 showed that both FVC and DLCO (FVC 101.8% predicted, DLCO 86.2% predicted) remained within normal limits. However, serial comparisons demonstrated mild declines in the percent predicted values of both parameters compared to previous measurements. CT revealed no significant disease progression since 2014 (Figure 1(b)). Genetic testing, performed with the patient’s consent, identified a homozygous mutation (c.910A>T) in exon 8 of the SLC34A2 gene (Figure 1(g)). A heterozygous mutation (c.910A>T) was found in his brother and son.
The patient exhibited mild symptoms, with daily activities not affected. The pulmonary function was normal, with no significant radiographic progression over the past decade. Given this clinical profile, no treatment was performed, and lung transplantation was deemed inappropriate. The patient was lost to follow-up after 1 month.
Case 2
The 44-year-old male developed a cough and sputum production in his teens. A chest X-ray at that time suggested alveolar microlithiasis. Regular CT scans showed increasing calcium deposition in both lungs in 2006. He did not receive treatment, and his dyspnea progressively worsened. He had no history of smoking or occupational dust exposure.
By October 2023, he presented at our hospital with worsening cough and dyspnea. Blood gas analysis indicated type I respiratory failure, with 90% oxygen saturation. PFTs revealed severe mixed ventilatory dysfunction (FVC 63.6% predicted, TLC 68.2% predicted, FEV1 49.2% predicted, and FEV1/FVC ratio 63.47%) and diffusion impairment (DLCO 44.5% predicted). Clubbing of the fingers was observed due to prolonged hypoxia. CT scans showed significantly increased pulmonary calcification compared to 2006, with additional calcifications in the aortic and coronary walls (Figure 1(c) and (d)). Ultrasound showed hepatic cysts and gallstones, but no abnormalities in the pancreas, spleen, or kidneys. Genetic testing confirmed a homozygous mutation (c.910A>T) in exon 8 of SLC34A2 (Figure 1(h)).
During hospitalization, bronchoscopy with BAL was performed, with next-generation sequencing of BAL fluid revealing Candida albicans infection. The patient received voriconazole antifungal therapy, supplemental oxygen, and acetylcysteine for expectoration. Given the patient’s reported maintenance of work capacity and CT findings showing partially preserved normal lung architecture. Based on this clinical presentation and imaging evidence, lung transplantation was not clinically indicated. The patient’s cough and sputum production improved after the treatment. Following discharge, home oxygen therapy was maintained. At the 1-month post-discharge follow-up, the patient maintained stable symptom control with oxygen saturation sustained above 90% without oxygen inhalation.
Case 3
The male patient was diagnosed with alveolar microlithiasis at 36 years of age in our outpatient clinic. Initially asymptomatic, he underwent regular follow-ups. Recently, he developed exertional dyspnea, intermittent cough, and sputum production. A CT scan showed disease progression. He was treated with antitussive agents and traditional Chinese medicine, which provided partial symptom relief. However, in September 2023, he presented with worsening symptoms. He had a long history of smoking, but had quit 3 years prior. He had a history of kidney stone surgery in 2020 (details unknown), and his family history was unavailable.
Blood gas analysis revealed a partial oxygen pressure of 75.6 mmHg and an SpO2 of 97%. PFTs demonstrated severe mixed ventilatory dysfunction (FVC 66% predicted, TLC 66.5% predicted, FEV1 48.4% predicted, and FEV1/FVC ratio 59.65%) and moderate diffusion impairment (DLCO 56.7% predicted). CT scans showed diffuse, punctate calcifications, primarily in the subpleural region, consistent with alveolar microlithiasis (Figure 1(e) and (f)). Echocardiography revealed moderate mitral valve insufficiency, possibly due to posterior mitral valve prolapse. Ultrasound of the liver, gallbladder, pancreas, spleen, kidneys, and ureters was normal. A biopsy of the basal segment of the right lower lobe revealed extensive calcification with chronic inflammatory cell infiltration in the subepithelial connective tissue. Genetic testing identified a homozygous mutation (c.575C>A) in exon 6 of SLC34A2 (Figure 1(i)).
Based on the patient’s symptoms and auxiliary examination findings, symptomatic treatments were administered: High-flow nasal cannula oxygen therapy, Levofloxacin as anti-infective therapy, corticosteroids, nebulization, and theophylline to relieve wheezing, and expectorant treatment. Despite presenting with wheezing and dyspnea, the patient maintained oxygen saturation above 95% with no significant impact on daily activities. Therefore, lung transplantation was not indicated. Subsequently, the patient’s symptoms improved, and he was discharged from the hospital. After discharge, the patient continued to take antitussive and expectorant medications, along with budesonide/glycopyrronium/formoterol inhalation aerosol. The patient reported stable symptoms but was lost to follow-up 1 month later.
Clinical and genetic characteristics of three new Chinese cases confirmed with genetic variations
The three new Chinese PAM patients have different clinical and genetic characteristics. All three patients had a long disease course, but with varying severity. PFTs indicated normal function in Case 1, while Cases 2 and 3 had severe ventilation impairment and moderate diffusion impairment. Imaging revealed minimal calcification in Case 1 over 10 years, whereas Case 2 exhibited progressive calcification, and Case 3 had diffuse calcification, reaching Radiographic Stage 4. Additionally, Cases 2 and 3 showed extrapulmonary calcifications. Overall, Case 1 had mild disease severity, while Cases 2 and 3 were moderate.
The mutation in cases 1 and 2, c.910A>T, is a nonsense mutation, located in the large EC-loop in NaPi-IIb, leading to premature termination of protein translation (p.Lys304*), and may also mediate mRNA decay to further decrease protein expression. The mutation in case 3, c.575C>A, is a missense mutation, substituting the 192nd amino acid from threonine to lysine (p.Thr192Lys). This missense mutation allows the protein to be translated normally, but it is located in the transmembrane domain 4, a critical functional domain of the protein. 55 To sum up, the severity of both variants is moderate according to the previously reported evaluation criterion. 11 The structural differences between normal SLC34A2 protein and the mutant proteins are illustrated in Figure 2.

Structural changes in sodium-dependent phosphate transport protein 2B caused by gene mutations of three patients compared to normal proteins. (a) Structure of the 304th amino acid (lysine) in normal protein. (b) The variant of c.910A>T (p.Lys304*) stops the translation of protein at the 304th amino acid. (c) Structure of the 192nd amino acid (threonine) in normal protein. (d) The variant of c.575C>A (p.Thr192Lys) causes the 192nd amino acid to be converted from Threonine to Lysine. Protein structure was predicted by SWISS-MODEL and drawn in PyMOL.
Results
The clinical manifestations in Chinese PAM patients with genetic identification
The study selection process and inclusion criteria are outlined in Figure 3. A total of 76 patients (73 patients from 36 reports, along with the 3 newly identified cases) were analyzed. Patients were categorized as Chinese or non-Chinese based on nationality.
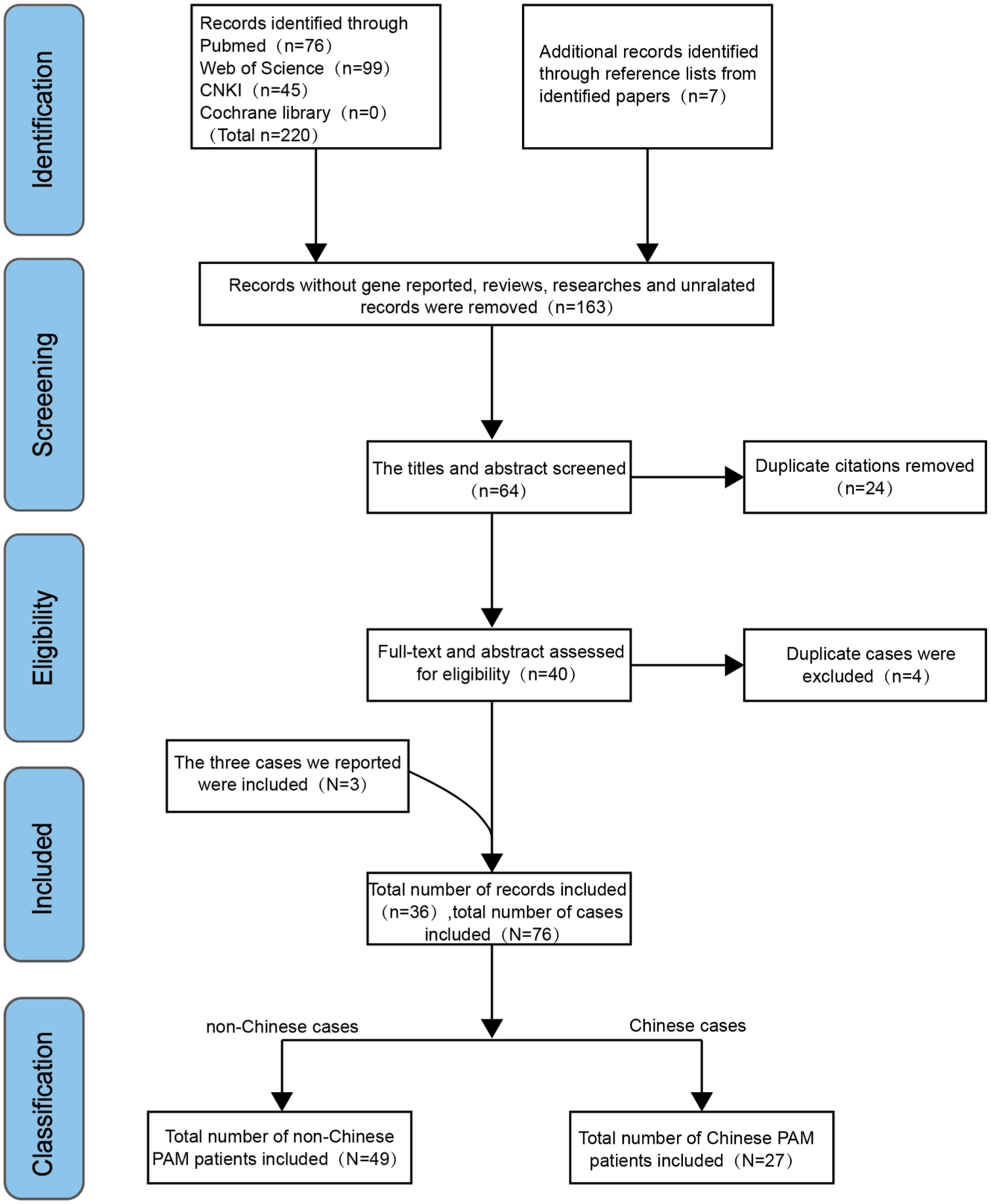
Flow chart of records and cases inclusion. Using “Pulmonary Alveolar Microlithiasis And SLC34A2” as keywords, reports containing PAM patients with SLC34A2 variants were included. After screening and exclusion, a total of 36 reports (n) were included, adding the 3 cases reported we mentioned, a total of 76 patients (N) were finally included. Classified by nationality, there were 27 Chinese and 49 non-Chinese PAM patients.
Since the SLC34A2 gene mutation was first identified as the cause of PAM in 2006,4,5 42 allelic mutations have been detected in at least 76 PAM patients, spanning exons 1–13 (Table 1, Figure 4). Some patients without detectable SLC34A2 mutations were excluded.56,57 One patient with PAM imaging features but only a single allele mutation was also excluded. 58 Another patient had imaging and pathological features consistent with microlithiasis, but a pathogenic mutation was found on only one allele; the mutation on the other allele (located in intron 2) did not affect the SLC34A2 coding sequence. Therefore, this case was not included in the analysis. 59
Summary of 76 patients carrying pathogenic SLC34A2 variants.
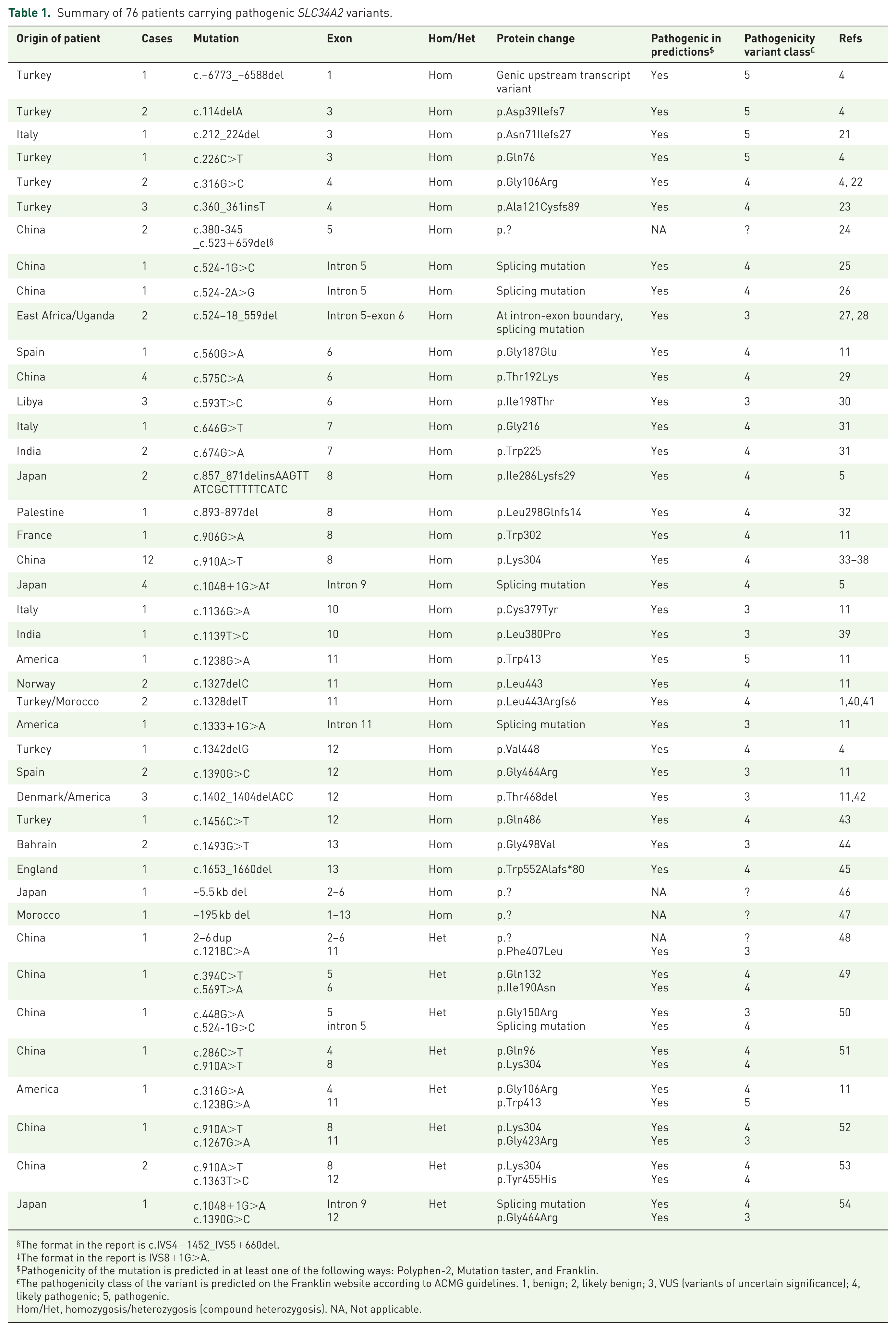
The format in the report is c.IVS4+1452_IVS5+660del.
The format in the report is IVS8+1G>A.
Pathogenicity of the mutation is predicted in at least one of the following ways: Polyphen-2, Mutation taster, and Franklin.
The pathogenicity class of the variant is predicted on the Franklin website according to ACMG guidelines. 1, benign; 2, likely benign; 3, VUS (variants of uncertain significance); 4, likely pathogenic; 5, pathogenic.
Hom/Het, homozygosis/heterozygosis (compound heterozygosis). NA, Not applicable.
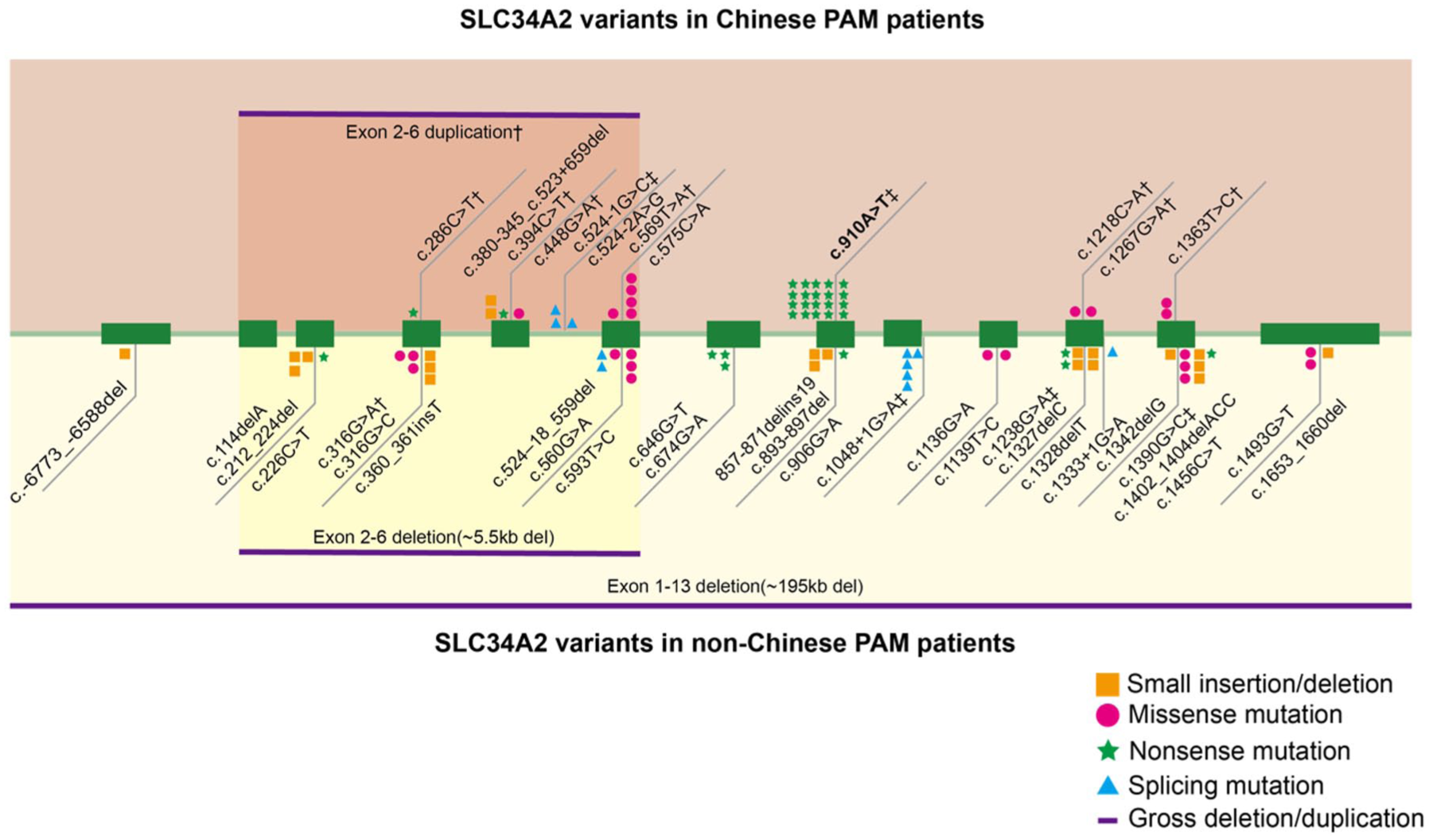
Comparison of SLC34A2 variants in Chinese and non-Chinese PAM patients. The green boxes represent exons. The orange area shows variants in Chinese patients, and the yellow area shows variants in non-Chinese patients. Different variant types are represented by different colored shapes. The number of different shapes represents the number of variants that occur. There are no common variants between the two populations. No special annotations represent homozygous variants.
A comparison of the demographic data and clinical manifestations between Chinese and non-Chinese PAM patients with SLC34A mutations is presented in Table 2. Among non-Chinese PAM patients with SLC34A2 mutations, 60.4% (29/48) were female, whereas 65.4% (17/26) of Chinese patients were male. The reported age of non-Chinese patients ranged from 1 to 69 years (median: 34 years; IQR: 17–45 years), while Chinese patients ranged from newborn to 57 years (median: 40 years; IQR: 33–51.25 years), exhibiting a narrower age range. More than half of both groups belonged to consanguineous families (58.1% and 58.3%, respectively).
Analysis of clinical characteristics of non-Chinese and Chinese PAM patients.

Data are presented as distributions (%), means ± SDs, or medians with IQRs. Analysis was performed using unpaired t-test, Pearson χ2 tests, Yates’s correction for continuity, Fisher’s Exact Test, or the Mann–Whitney U test.
DLCO, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; IQR, interquartile range; PAM, pulmonary alveolar microlithiasis.
n: indicates the number of valid cases available for analysis for that specific clinical characteristic.
There was no significant difference in clinical symptoms between the groups. Some patients remained asymptomatic (25.7% in non-Chinese and 20.0% in Chinese patients). Dyspnea (60% vs 68%) and cough (48.6% vs 52%) were the most common symptoms. A few patients exhibited sputum production (2.9% vs 16%), chest pain (14.3% vs 8%), and fatigue (22.9% vs 4%). Mortality rates were 11.9% in non-Chinese patients and 0% in Chinese patients.
Comparison of SLC34A2 variant spectra between non-Chinese and Chinese PAM patients
Most of the mutations observed were homozygous, with compound heterozygous mutations primarily found in Chinese patients. Among 49 non-Chinese PAM patients, 47 (95.9%) had homozygous mutations in SLC34A2, whereas 20 out of 27 Chinese patients (74.1%) had homozygous mutations. Among the homozygous mutations, deletions/insertions were most common in non-Chinese patients (19/47, 40.4%), while nonsense mutations were the most frequent in Chinese patients (12/20, 60%). A detailed comparison of the genetic characteristics of 76 patients is summarized in Table 1.
Our data reveal significant differences in the SLC34A2 mutation spectrum between non-Chinese and Chinese patients (Figure 4). Variants found in Chinese patients are absent in non-Chinese patients, and vice versa. The c.910A>T (p.Lys304*) mutation is the most common variant in China, identified in 16 patients from 13 unrelated families. The second most common variant is c.575C>A (p.Thr192Lys), observed in four patients from two unrelated families. Both variants are restricted to Chinese patients. Common variants in Chinese PAM patients are concentrated in exons 5, 6, and 8, while in non-Chinese patients, mutations span exons 1–13 of the SLC34A2 gene.
Of the 42 variants detected, 34 (81%) were homozygous, while 8 (19%) were compound heterozygous. Among the Chinese patients, 7 had 6 different heterozygous combinations (including c.2-6dup and c.1218C>A, c.394C>T and c.569T>A, c.448G>A and c.524-1G>C, c.286C>T and c.910A>T, c.910A>T and c.1267G>A, c.910A>T and c.1363T>C). In contrast, only two non-Chinese patients had heterozygous combinations (c.316G>A and c.1238G>A, c.1048+1G>A, and c.1390G>C). These compound heterozygous mutations involve at least one variant that affects the coding sequence of SLC34A2 (e.g., long segment variants, splicing mutations, and nonsense mutations). Five of these variants (c.524-1G>C, c.910A>T, c.1048+1G>A, c.1238G>A, c.1390G>C) have been previously reported as homozygous mutations.
Eleven allelic variants (11/42, 26.2%) were found in China, making it the country with the highest number of SLC34A2 variants, followed by Turkey (8/42, 19%), Japan (4/42, 9.5%), and the United States (4/42, 9.5%). Almost all allelic variants have been reported in only one country, but three variants (c.524–18_559del, c.1328delT, c.1402_1404delACC) were described in patients from two countries, suggesting that the SLC34A2 variation may exhibit some regional hotspots.
Comparison of clinical manifestations among patients with different SLC34A2 variant types
To further explore differences in clinical manifestations across SLC34A2 mutation types, we classified the 67 patients with homozygous mutations into four categories based on mutation type: Deletion/Insertion, Missense, Nonsense, and Splicing mutations. Of the 52 patients with available clinical data, more than half of those carrying missense mutations were asymptomatic (7/12, 58.3%), whereas all patients with nonsense or splicing mutations exhibited respiratory symptoms (19/19, 100% and 5/5, 100%, respectively; p < 0.001). Furthermore, dyspnea was present in the vast majority of patients with nonsense mutations (16/19, 84.2%), compared to only a minority of those with missense mutations (4/12, 33.3%; p = 0.034; Table 3). These results demonstrate a statistically significant genotype-phenotype correlation. In addition, I compared the clinical information of Chinese patients with the c.910A>T homozygous mutation and other mutations. There was no significant difference. However, patients with this mutation all had respiratory symptoms, while some patients (3/8, 37.5%; p = 0.058) without this mutation were asymptomatic (Supplemental Table 2).
Analysis of clinical characteristics of different SLC34A2 mutation types.
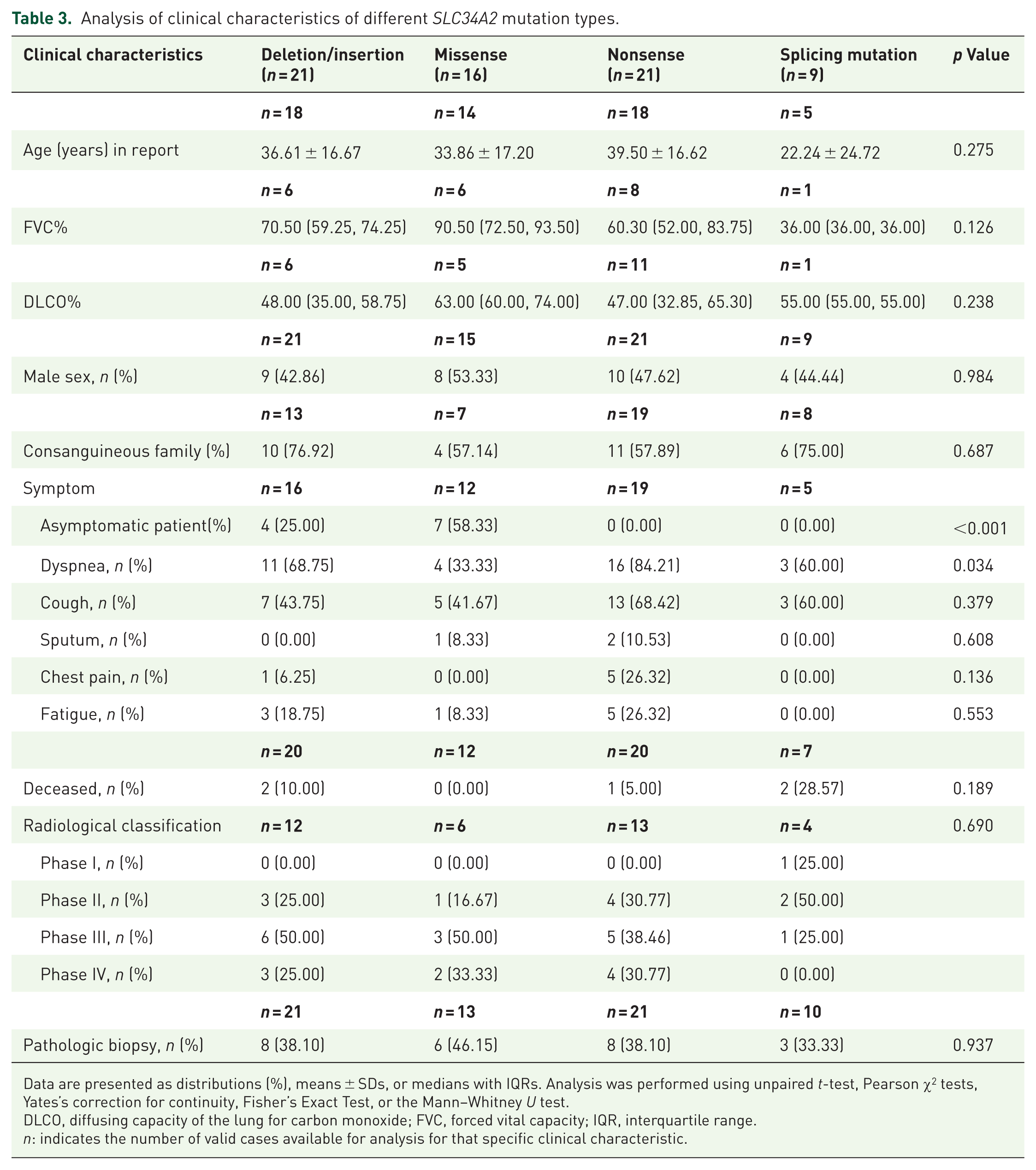
Data are presented as distributions (%), means ± SDs, or medians with IQRs. Analysis was performed using unpaired t-test, Pearson χ2 tests, Yates’s correction for continuity, Fisher’s Exact Test, or the Mann–Whitney U test.
DLCO, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; IQR, interquartile range.
n: indicates the number of valid cases available for analysis for that specific clinical characteristic.
Discussion
Genetic analysis showed that Case 2 and Case 3 harbored SLC34A2 mutations of comparable severity, which explains their similar clinical manifestations. This observation is consistent with the established genotype-phenotype correlation principle. Interestingly, despite sharing the same genetic variant, Case 1 and Case 2 exhibited different clinical severity. This discrepancy suggests that other factors, such as environmental influences, epigenetic modifications, and co-mutations, may also impact clinical manifestations. Larger cohorts are needed to investigate the influence of multiple factors on the onset and progression of PAM.
Among the cases we reported, none of the three patients received specific treatment. Currently, there are no established treatment guidelines for PAM. Corticosteroids, calcium-binding agents, and bronchoalveolar whole lung lavage are generally considered ineffective. 16 A low-phosphate diet and therapeutic EDTA lavage reduced microliths deposited in the lungs of PAM mice, but their efficacy remains unconfirmed in humans. 60 Bisphosphonates bind to bone minerals (hydroxyapatite) with high affinity, inhibiting osteoclast activity and osteoclast-mediated bone resorption. Only a few patients treated with bisphosphonates showed clinical and radiographic improvement, 4 while others remained unresponsive.42,61 More clinical data are needed to support its therapeutic effect. Lung transplantation is considered the only effective treatment so far. 16 A total of 29 PAM patients who underwent lung transplantation were reported among the 22 publications. Twenty-three out of 29 received a double lung transplant, and 6 received a single lung transplant. As for the prognosis, 18 out of 25 patients survived, and the symptoms and lung function of these survivors improved. The causes of death of the deceased patients include obliterative bronchiolitis (two cases), sepsis (two cases), hemodynamic instability (one case), and multiorgan failure (two cases).62–83 However, lung transplantation is costly and not all end-stage PAM patients can receive treatment. Gene-editing techniques, as highly focused gene therapies such as CRISPR-Cas9 and CRISPR-Cas13, may offer future therapeutic potential for repairing SLC34A2 mutations. Additionally, mRNA-based protein replacement therapy can be used to treat monogenic diseases by supplementing deficient proteins. In vitro-transcribed SLC34A2 mRNA could represent a potential therapy for PAM.
Establishing a genotype-phenotype correlation is crucial for personalized treatment strategies, improving effectiveness, and minimizing side effects. In our study, a slight male predominance was observed among Chinese PAM patients, while a higher proportion of female patients was noted in non-Chinese populations. These findings align with previously reported epidemiological patterns. 84 In addition, Chinese PAM patients frequently exhibited multiple compound heterozygous mutations, likely due to the large population base and a preference for non-consanguineous marriage (Supplemental Table 3). Genetic analysis of the mutation spectrum indicates that the nonsense c.910A>T mutation in exon 8 was most prevalent among homozygous mutations, representing a potential screening target for Chinese PAM patients. 34 Variants in exons 5 and 6 were also common. In contrast, non-Chinese patients exhibited a higher prevalence of deletion and insertion mutations. The strikingly different mutation spectra between Chinese and non-Chinese patients may reflect ancestral genetic divergence and environmental influences. Identifying these genetic patterns will aid early diagnosis and guide personalized treatment strategies.
Despite different variant spectra, clinical manifestations and imaging findings were comparable between Chinese and non-Chinese patients. This phenotypic similarity may be attributed to the similar functional consequences of both deletion/insertion mutations and nonsense mutations. Deletion/insertion mutations induce frameshifts that distort protein structure, whereas nonsense mutations truncate the polypeptide chain. Despite these distinct mechanisms, both ultimately compromise protein function. Notably, significant clinical variability was observed across different mutation types, suggesting genotype-phenotype correlations in PAM. In addition, some PAM patients lack detectable SLC34A2 mutations,56,57 suggesting alternative pathogenic mechanisms. A recent Chinese case identified an FBN1 mutation in a PAM patient. 10 Further investigation into other potential genetic contributors, such as genes affecting calcium-phosphorus metabolism, epigenetic regulation, environmental influences, and multifactorial inheritance, is essential for improving PAM diagnosis and treatment.
However, this study has several limitations. First, the analysis was restricted to patients with reported SLC34A2 mutations, and the relatively small number of cases may introduce statistical bias. In addition, incomplete information and inconsistent radiological classification due to reporting at different disease stages may affect accuracy. Future longitudinal studies are necessary for a better understanding of disease progression.
Conclusion
In this study, we report the clinical course of three new PAM patients and review the most recent characteristics of SLC34A2 gene mutations in Chinese PAM patients. Comparison of genetic characteristics between Chinese and non-Chinese PAM patients shows that nonsense variants are the most common in Chinese patients, with c.910A>T being a unique gene target in this population.
Supplemental Material
sj-docx-1-tar-10.1177_17534666251381679 – Supplemental material for Differences in genetic characteristics between Chinese and non-Chinese patients with pulmonary alveolar microlithiasis—case series and a systematic review
Supplemental material, sj-docx-1-tar-10.1177_17534666251381679 for Differences in genetic characteristics between Chinese and non-Chinese patients with pulmonary alveolar microlithiasis—case series and a systematic review by Mengyao Guo, Lijuan Hua, Wenxue Bai, Xuezhao Wang, Dongyuan Wang, Lirong Chen, Bingyi Liu and Min Xie in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.