
Abstract
Restrictive cardiomyopathy (RCM) is a rare childhood cardiomyopathy that is a challenging diagnostic problem for clinicians. We describe a case of an 8-year-old girl with a 2-year history of shortness of breath on exertion. Electrocardiogram and echocardiography showed biatrial enlargement, while cardiac magnetic resonance showed biatrial dilation and normal pericardial thickness. Left and right heart catheterization revealed a left ventricular (LV) end-diastolic pressure (EDP) of 20 mmHg, right ventricular (RV) EDP of 13 mmHg, and pulmonary arterial systolic pressure of 51 mmHg. LV and RV pressure traces showed that LV and RV pressures moved concordantly with respiration, and that the systolic area index was 0.98. Cardiac catheterization data were therefore supportive of RCM. Next-generation sequencing identified a heterozygous variant of the troponin I gene (TNNI3; c.574C>T). Combining these findings led to a diagnosis of RCM. The patient’s parents chose conservative treatment, but at the 12-month follow-up she died of worsening heart failure and cerebral infarction. This case emphasizes the need for cardiac catheterization and genetic testing in RCM, and suggests that anticoagulants should be recommended to reduce the risk of thromboembolic events.
Introduction
Pediatric cardiomyopathies are a heterogeneous group of rare disorders, of which dilated and hypertrophic cardiomyopathy are the most common types, while left ventricular (LV) noncompaction cardiomyopathy and restrictive cardiomyopathy (RCM) have a lower frequency. 1 , 2 Indeed, RCM accounts for only 2% to 5% of all pediatric cardiomyopathies. It is defined as diastolic ventricular dysfunction with enlarged left and right atria, preserved LV systolic function, and normal ventricular size and wall thickness. 3 , 4
RCM is a challenging diagnostic problem for clinicians because it can be difficult to differentiate from constrictive pericarditis (CP). Cardiac catheterization can help identify the typical hemodynamic responses of RCM and CP,5–7 while mutations in sarcomere and sarcomere-related genes have been identified in patients with RCM.8–11 Here, we report a rare case of pediatric RCM that was diagnosed by a combination of cardiac catheterization and genetic analysis.
Case report
The reporting of this study conforms to CARE guidelines. 12 An 8-year-old girl with a 2-year history of progressive shortness of breath on exertion was admitted to our hospital. She had no history of hypertension, diabetes, tuberculosis, connective tissue disorders, or radiotherapy, and no family history of sudden cardiac death or cardiomyopathy. She was diagnosed with heart failure and received intermittent diuretic therapy.
Physical examination revealed mild edema of both lower limbs. An electrocardiogram showed biatrial enlargement of ST-segment depression in the inferior and lateral precordial leads (Figure 1). Echocardiography revealed the enlargement of both atria, nondilated ventricular chambers, an interventricular septum and ventricular wall of normal thickness, slight pericardial effusion, and a LV ejection fraction of 55% (Figure 2A). The pulsed-wave Doppler mitral inflow pattern and tissue Doppler imaging showed evidence of restrictive filling: the ratio of early to late peak velocities (E/A) was 3.12, and the ratio of the transmitral E-wave to e′ velocity (E/e′ ratio) was 15.56 after averaging the e′-wave velocities from the septal and lateral points of the mitral annulus (Figure 3). The LV global longitudinal strain measured by speckle-tracking echocardiography was normal. Cardiac magnetic resonance showed remarkable biatrial enlargement, but normal pericardial thickness (Figure 2B). Late gadolinium enhancement was negative. Coronary computed tomography (CT) angiography revealed a normal coronary artery. Cardiac and pulmonary CT angiography showed no evidence of pulmonary embolism, anomalous pulmonary venous drainage, or pulmonary arteriovenous fistulas. Laboratory evaluation revealed a significant increase in serum N-terminal pro-B-type natriuretic peptide levels (2820 ng/L; normal range: 0–125 ng/L). The following laboratory findings were normal: thyroid function, liver chemistry tests, rheumatic disease antibody levels, troponin I, creatinine, and blood urea nitrogen levels, white blood cell count, urine analysis, D-dimer levels, hepatitis panel screening, and serum free light chain concentrations; HIV antibody levels were negative.

Electrocardiogram showing biatrial enlargement of ST-segment depression in the inferior and lateral precordial leads

(a) Echocardiography showing enlargement of both atria. Right atrium: long axis diameter, 3.71 cm; short axis, 3.41 cm. (b) Cardiac magnetic resonance imaging showing enlargement of both atria

(a) Pulsed-wave Doppler mitral inflow filling pattern showing a high E/A ratio. (b) Tissue Doppler imaging showing e′-wave velocities from the septal wall of the mitral annulus. (c) Tissue Doppler imaging showing e′-wave velocities from the lateral wall of the mitral annulus
Invasive left and right heart catheterization showed a cardiac index of 2.35 L/minute/m2, a LV end-diastolic pressure (LVEDP) of 20 mmHg, right ventricular end-diastolic pressure (RVEDP) of 13 mmHg, right ventricular systolic pressure (RVSP) of 55 mmHg, pulmonary arterial systolic pressure (PASP) of 51 mmHg, RVEDP/RVSP of 0.236, and pulmonary vascular resistance of 5.12 Wood units. LV and RV pressure traces revealed that LV and RV pressures moved concordantly with respiration (Figure 4), and the systolic area index was 0.98. These hemodynamic data suggested RCM.
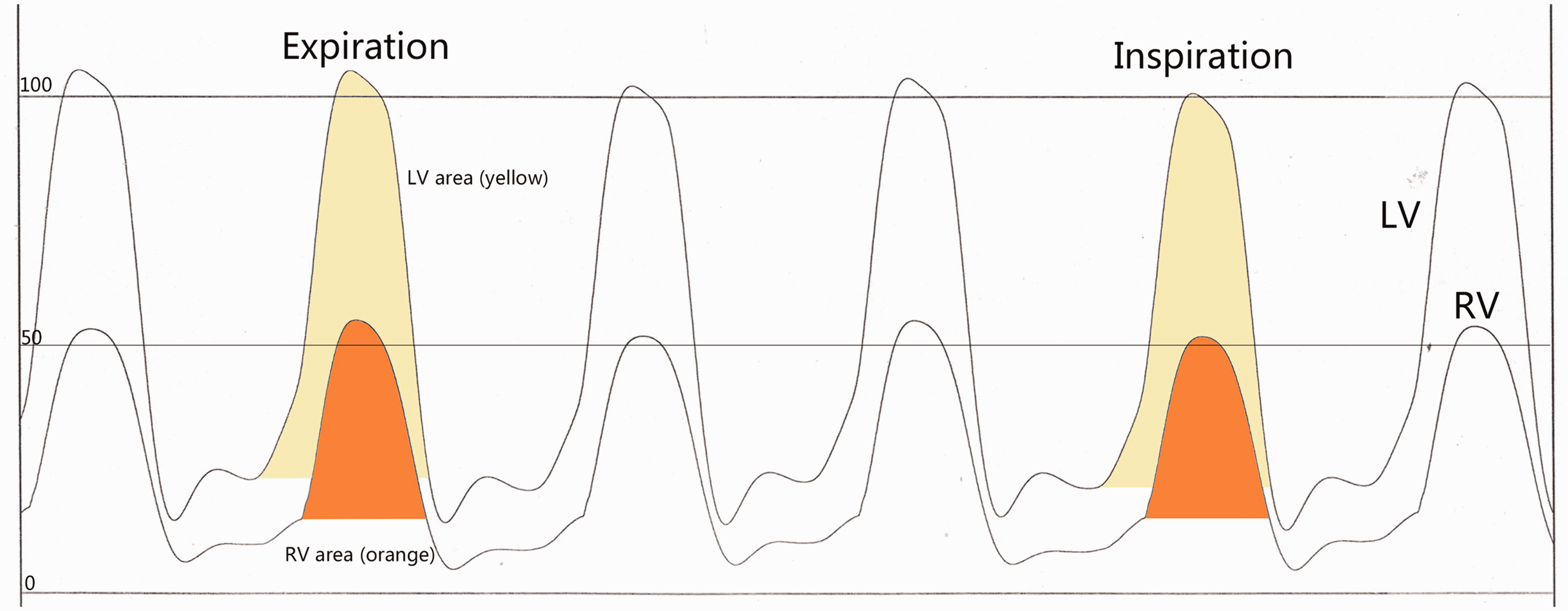
LV and RV pressure traces showing that LV and RV pressures move concordantly with respiration. Orange shading represents the area of the RV pressure curve; yellow shading represents the area of the LV pressure curve
Several genes have been implicated in RCM. Although the patient had no family history of sudden cardiac death or cardiomyopathy, genetic analysis was performed by exome sequencing. A heterozygous variant of the troponin I gene (TNNI3; NM_000363, c.574C > T p.R192C) was identified in the patient, which was absent in her parents (Figure 5); it has previously been reported in cases of RCM.13–15 Therefore, the combined results of cardiac catheterization and genetic analysis led to a diagnosis of RCM. Her parents did not accept our recommendation of a heart transplantation, and her symptoms were relieved by diuretics treatment. However, at the 12-month follow-up, the patient died of worsening heart failure and cerebral infarction.

Family tree and the TNNI3 gene. (a) Family pedigree of the proband (black arrowhead). Circle: female, square: male. (b) Analysis of exome sequencing data confirming a heterozygous TNNI3 mutation. Red arrowhead indicates the point of variation for p.R192C (c.574C > T). The chromatogram shows A/G rather than C/T because Sanger sequence analysis is in the reverse direction
Discussion
Pediatric RCM is a rare disorder with a large heterogeneous range of causes. The differentiation of CP from RCM is challenging because the two conditions share a common clinical manifestation, mainly right heart failure, and similar echocardiographic findings. However, the treatment options are different, with pericardiectomy shown to be effective in CP, so accurate differentiation is crucial. 5 , 16
CT and magnetic resonance imaging can be used to assess the pericardial anatomy, but these tests do not necessarily reveal pathophysiological abnormalities. Moreover, although a pathologically thickened pericardium (>4 mm) is highly suggestive of CP, this finding should not be used alone because some patients with surgically proven CP have a normal pericardial thickness. 6 Additionally, patients may have a thickened pericardium without constriction, especially after cardiac surgery or radiation therapy. 99mTechnetium-pyrophosphate scintigraphy is a useful clinical tool for the diagnosis of transthyretin cardiac amyloidosis, 17 but it was not performed in this case because other parameters (such as a normal wall thickness, the global longitudinal strain, late gadolinium enhancement, and serum free light chain concentrations) and the young age of the patient did not support a diagnosis of cardiac amyloidosis.
Cardiac catheterization can help identify typical hemodynamic responses of RCM and CP.5–7 Conventional catheterization criteria for RCM include severe pulmonary hypertension (PASP >50 mmHg) and a large difference between LVEDP and RVEDP (LVEDP – RVEDP >5 mmHg). Respirophasic variation in pressures is the most useful hemodynamic catheterization finding to distinguish RCM from CP. The systolic area index [defined as the ratio of the RV area (mm Hg × s) to LV area (mm Hg × s) in inspiration vs. expiration] has both good sensitivity and specificity for the diagnosis of CP (cutoff >1.1, 97% sensitivity, 100% specificity). 6 In our patient, the hemodynamic data of left and right heart catheterization suggested RCM.
Several genes have previously been implicated in RCM, including MYH7, TNNI3, TNNT2, MYL2, MYL3, ACTC, MYBPC3, TPM1, and DES. 4 , 18 We used exome sequencing to identify a heterozygous variant of TNNI3 (c.574C > T p.R192C) in this patient, which has already been reported in RCM. 13–15
Currently, the treatment of RCM is mainly symptomatic, and is largely limited to the use of diuretics in patients with signs and symptoms of systemic or pulmonary venous congestion. Drug therapy has not shown any significant long-term benefits, and heart transplantation is the only effective therapy. 19 Patients with enlarged atria are at an increased risk of forming blood clots, so patients with RCM should be recommended anticoagulants to reduce the risk of this.
In conclusion, we report a rare case of pediatric RCM. This case emphasizes the need for cardiac catheterization and genetic testing in RCM, and for the recommendation of anticoagulants to reduce the risk of thromboembolic events.
Footnotes
Acknowledgements
We thank Ji-Wang Chen, PhD, Section of Pulmonary, Critical Care Medicine, Sleep and Allergy, Department of Medicine, University of Illinois at Chicago, Chicago, IL, USA, for editing and revising the English text of this manuscript.
Author contributions
XFG and HLD collected the data and participated in writing the manuscript. HLD conceived the study and participated in writing the manuscript. HLD, and XFG participated in patient management. QHW, XS, and YCD participated in patient echocardiography examination. All authors contributed to the article and approved the submitted version.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics statement and informed consent
Written consent for treatment, genetic testing, and publication were obtained from the guardian of the patient, and written consent for publication was obtained from a legally authorized representative. Ethics approval was not required because this was a case report.
Funding
This study was supported by the National Natural Science Foundation of China (82060018, 81700438), and Yunnan Fundamental Research Projects (202101AS070043, 202102AA310003-7, 202105AF150019, and 202301AY070001-300).