
Abstract
Background:
In patients with chronic fibrosing interstitial lung disease (ILD), some may develop a progressive fibrosing (PF) phenotype, which presents as rapid progression and often results in poor clinical outcomes.
Objective:
The objective of this study was to construct and test a model to identify independent predictors of mortality in PF-ILD and to trace the lung function trajectory of patients with PF-ILD.
Design:
This multicenter retrospective cohort study enrolled patients with PF-ILD from two distinct centers with 8-year follow-up to develop and validate a prognostic nomogram based on clinical factors and assess longitudinal lung function trajectories.
Methods:
We enrolled patients diagnosed with PF-ILD from China-Japan Friendship Hospital (training cohort) and Jiulongpo Hospital of Traditional Chinese Medicine (validation cohort). Survival status was recorded during the 8-year follow-up period. Clinical demographics, laboratory data, pulmonary function test (PFT) results, and high-resolution computed tomography results were collected for analysis. A training cohort of patients with PF-ILD was used to identify predictors of mortality, which were then validated in an external cohort. A nomogram was established based on multivariate factors. The predictive performance of the model was evaluated using receiver operating characteristic curves and calibration curves. Survival estimates were performed using the Kaplan–Meier method and compared using the log-rank test. The PFT trajectory was estimated using a linear mixed model.
Results:
A total of 1419 patients with PF-ILD from China-Japan Friendship Hospital (training cohort) and 282 patients with PF-ILD from Jiulongpo Hospital of Traditional Chinese Medicine (validation cohort) were enrolled. During the 8-year follow-up, 150 (10.57%) patients received lung transplantation, while 43.55% (n = 618) of cases reached mortality, with a median survival time of 53 months in the BJ cohort. A predictive model was built based on ILD subgroups, baseline Forced Vital Capacity%pred (FVC%pred), baseline Diffusing Capacity of the Lung for Carbon Monoxide%pred (DLCO%pred), age at diagnosis, antifibrosis treatment, gastroesophageal reflux complication, C-Reactive Protein levels, and BMI. We also found AEs and progressive declines in FVC and DLCO, particularly after the third year post-diagnosis, were strongly associated with poor prognosis and may serve as important longitudinal biomarkers for risk stratification in PF-ILD.
Conclusion:
A predictive model incorporating multiple factors effectively predicted 8-year survival in patients with PF-ILD. In addition to these baseline predictors, AEs and progressive declines in FVC and DLCO were strongly associated with poor prognosis and may serve as valuable longitudinal biomarkers for ongoing risk stratification.
Plain language summary
Some patients with chronic interstitial lung disease (ILD) develop a progressive form (PF-ILD) that worsens quickly and leads to poor outcomes. In this study, we followed over 1,700 PF-ILD patients from two hospitals for eight years to identify factors linked to survival.
We created a model using information like lung function, age, treatments, and other health conditions to predict which patients had higher risk of death. We found that poor lung function at diagnosis, certain complications, and older age were linked to worse outcomes. In addition, patients who had acute exacerbations (AEs) or showed lung function decline after the third year had a higher risk of death.
This model can help doctors monitor PF-ILD patients more closely and provide earlier intervention.
Introduction
Interstitial lung diseases (ILDs) comprise a heterogeneous group of pulmonary disorders characterized by varying degrees of inflammation and fibrosis. 1 Among patients with non-idiopathic pulmonary fibrosis (non-IPF) forms of chronic fibrosing ILDs, a subset may develop a progressive disease course, manifesting as worsening respiratory symptoms, accelerated decline in lung function, and increased early mortality—despite receiving conventional treatment. This clinical behavior closely resembles that of idiopathic pulmonary fibrosis (IPF) and is now referred to as progressive fibrosing interstitial lung disease (PF-ILD).2,3 To standardize the identification of PF-ILD, several consensus definitions have been proposed. These commonly include criteria such as a categorical decline in lung function, radiological progression of fibrosis on high-resolution chest imaging, symptomatic deterioration, or a combination of these features.4–6 Epidemiological studies from various countries estimate that approximately 14%–32% of patients with non-IPF ILDs may exhibit a progressive fibrosing phenotype. 7
The INBUILD trial demonstrated that nintedanib effectively attenuates disease progression in a heterogeneous population of patients with PF-ILD, primarily by slowing the decline in forced vital capacity (FVC). 4 Based on these findings, nintedanib has received regulatory approval in the United States and over 45 other countries for the treatment of chronic fibrosing ILDs with a progressive phenotype. Additionally, the RELIEF study evaluated the efficacy of pirfenidone in patients with progressive pulmonary fibrosis and reported a significant reduction in early disease progression. However, the trial was prematurely terminated due to inadequate patient enrollment. 6 Both nintedanib and pirfenidone exhibit antifibrotic and anti-inflammatory properties that contribute to slowing ILD progression. Nevertheless, neither agent has shown a significant impact on overall survival. It is well established that patients with PF-ILD experience significantly lower long-term survival compared to those without a progressive phenotype.8,9 The intrinsic heterogeneity of PF-ILD presents a substantial challenge in assessing risk factors associated with long-term survival. To address this, clinical prediction models have been developed for specific ILD subtypes, such as IPF, 10 autoimmune-related ILD,11,12 and sarcoidosis. 13 However, for the broader PF-ILD population, the prognostic value of baseline demographic, functional, and radiographic factors remains unclear. Furthermore, most therapeutic trials have utilized short-term endpoints—particularly FVC decline—as the primary measure of efficacy.4,14 Due to limitations in follow-up duration, long-term survival outcomes are seldom employed as primary endpoints in clinical trials. In this study, we aimed to develop a clinical prediction model for survival in patients with PF-ILD by integrating comprehensive clinical variables and identifying key determinants of mortality through long-term follow-up analysis.
Methods
Subjects
Data from patients with ILD who were hospitalized at the China-Japan Friendship Hospital and Jiulongpo Hospital of Traditional Chinese Medicine between October 2022 and July 2024 were retrospectively retrieved. All patients met the diagnostic criteria for ILD as defined by the American Thoracic Society (ATS), the European Respiratory Society (ERS), or the official ATS/ERS/Japanese Respiratory Society/Latin American Thoracic Society statement.15,16 ILD diagnoses were confirmed by the treating specialist, and in cases of diagnostic uncertainty, a multidisciplinary review was conducted by chest radiologists and, when necessary, lung pathologists. The classification of ILD patients was determined according to established consensus guidelines and previously published literature.2,15–21
Disease progression was evaluated using overlapping windows of 24 months prior to each hospital visit. Progression was confirmed when the first event meeting the predefined criteria for disease progression was observed. The date of “diagnosis” was defined as the first occurrence of criteria fulfillment for PF-ILD. The end of follow-up was determined as the date of death or the end of the study period. Survival duration was calculated from the “diagnosis” date to the date of death or the end of the study.
The following parameters were applied for the definition of PF-ILD during time in the registry:20,21 ⩾10% relative decline in FVC; 5%–9% relative decline in FVC with worsening respiratory symptoms; 5%–9% relative decline in FVC with CT progression of fibrosis; 5%–9% relative decline in FVC with ⩾15% relative decline in diffusing capacity of the lung for carbon monoxide (DLCO); and CT progression of fibrosis with worsening respiratory symptoms.
Patients were excluded if they met any of the following criteria 4 : (1) Alanine transaminase, aspartate transaminase, or total bilirubin levels > 1.5 times the upper limit of normal. (2) Creatinine clearance < 30 mL/min, as calculated by the Cockcroft–Gault formula. (3) Chronic hepatic disease (Child-Pugh A, B, or C). (4) Bleeding risk (defined as genetic predisposition to bleeding; requirement for fibrinolysis, full-dose therapeutic anticoagulation, or high-dose antiplatelet therapy; history of hemorrhagic central nervous system event within 12 months; hemoptysis or hematuria, active gastrointestinal bleeding, or gastrointestinal ulcers; or major injury or surgery within 3 months; or international normalized ratio > 2, prolongation of prothrombin time, and activated partial thromboplastin time > 1.5 times the upper limit of normal) at screening. (5) Significant pulmonary arterial hypertension (PAH), defined as previous clinical or echocardiographic evidence of significant right heart failure; history of right heart catheterization showing a cardiac index ⩽2 L/min/m2. (6) History of severe uncontrolled hypertension (⩾160/100 mmHg) within 6 months, history of myocardial infarction within 6 months, history of unstable angina within 6 months, or history of thrombotic event (including stroke and transient ischemic attack) within 12 months of screening. (7) Clinically diagnosed malignancies in any system.
Study design
This multicenter study established distinct cohorts for model development and validation. The training cohort consisted of patients diagnosed with PF-ILD at the China-Japan Friendship Hospital, while the validation cohort included patients from Jiulongpo Hospital of Traditional Chinese Medicine. Baseline data, defined as the date on which patients first met the criteria for PF-ILD, were collected from medical records. These data included clinical, laboratory, radiographic, cytological, and pathological information. Survival status was obtained from medical records or confirmed through telephone interviews with patients or their families. Longitudinal pulmonary function test (PFT) data, including FVC and DLCO, were collected at each hospital visit, from baseline until the end of the study period. In addition, adverse events (AEs) were assessed during follow-up. AEs of ILD were defined according to the latest international consensus 22 as acute, clinically significant respiratory deteriorations meeting all of the following criteria: (1) new or worsening dyspnea within less than 1 month of onset; (2) high-resolution computed tomography (HRCT) revealing new bilateral ground-glass opacities or consolidation superimposed on preexisting fibrotic abnormalities; and (3) absence of an alternative explanation such as cardiac failure or fluid overload. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement, 23 details were provided in S Table 1.
Statistical analysis
Demographic and clinical characteristics of survivors and non-survivors were compared. Continuous variables were described based on their distribution patterns: parametric data were presented as mean ± standard deviation (SD), nonparametric variables as median (interquartile range (IQR)), and categorical variables as frequency percentages. Intergroup comparisons were performed using the following tests: student’s t-test for Gaussian-distributed variables, Mann–Whitney U-test for skewed distributions, and χ2 or Fisher’s exact tests for contingency analyses.
Exploratory multivariate analyses were conducted using a Cox proportional hazards model to identify factors associated with mortality. The model incorporated baseline clinical, laboratory, radiographic, cytological, and pathological data. Continuous predictors were categorized based on prior studies,11,21,24 with optimal cutoff values determined through area under the curve (AUC) analysis. Prognostic factors significantly associated with overall survival in univariate analyses (p < 0.001) were included in the multivariate model. A nomogram was developed based on the results of the multivariate analysis (p < 0.001) using a training cohort of patients with PF-ILD. Overall survival was defined as the time from the first fulfillment of PF-ILD inclusion criteria to death from any cause or the end of the follow-up period. Antifibrosis treatment was defined as the regular use of antifibrotic agents for more than 3 months after the diagnosis of ILD and before meeting the PF-ILD criteria. Concomitant therapies were defined as the use of corticosteroids, mycophenolate mofetil, cyclophosphamide, azathioprine, methotrexate, or rituximab. 25 To evaluate the potential confounding effect of concomitant therapies on the association between antifibrotic treatment and mortality, a sensitivity analysis was performed. Specifically, we repeated the Cox regression analysis after excluding patients who received concomitant therapies to ensure that the observed survival benefit of antifibrotic treatment was independent of these additional interventions. The predictive performance of the models was evaluated using receiver operating characteristic (ROC) curves and calibration curves. External validation was performed using an independent validation cohort. Survival estimates were calculated using the Kaplan–Meier method for the overall population and compared between the training and validation cohorts using the log-rank test.
PFTs were analyzed using a linear mixed-effects model, applied to raw measurements. This model incorporated a simple random-effect intercept and ILD subgroup slope, with fixed-effect intercept and slope terms, and an unstructured G-matrix covariance. Given the variability in time intervals between measurements across subjects, standardized time points were set at 12-month intervals. PFT measurements were assigned to these time points using a ±3-month window, selecting the nearest available measurement for analysis.
Results
A total of 4762 hospitalized patients with ILD were screened between October 2016 and July 2024. Specifically, 3795 patients were from the China-Japan Friendship Hospital, and 967 patients were from Jiulongpo Hospital of Traditional Chinese Medicine. A flowchart outlining the patient screening process is presented in S Figure 1.
Patient demographics and baseline characteristics
In the BeiJing (BJ) cohort, the mean age was 59.86 years, and 53.07% (n = 753) of patients were male. The mean FVC% pred and DLCO% pred were 76.96% and 57.90%, respectively. Among the cohort, 39.30% (n = 558) were on antifibrotic therapy, with 34.83% (n = 495) receiving pirfenidone and 4.44% (n = 63) receiving nintedanib (Table 1). The etiologies of ILD in the BJ cohort included autoimmune ILDs (37%), non-IPF idiopathic interstitial pneumonia (IIP) (27.48%), IPF (17.12%), sarcoidosis (9.73%), and exposure-related ILD (8.67%) (S Table 2).
Descriptive statistics of PF-ILD cohort and comparison between survivors and non-survivors in training cohort.

Statistically significant subgroup effect sizes were ascertained as P subgroup (χ2 test) < 0.001. Data are presented as numbers (%) or mean ± standard deviation. Pulmonary function data were expressed in liters and as a percentage of the predicted (% pred).
CRP, C-reactive protein; DLCO, diffusion capacity of carbon monoxide; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; PCO2, partial pressure of carbon dioxide; PO2, partial pressure of oxygen.
Comparison of characteristics between survivors and non-survivors
In the BJ cohort, at the time of meeting the diagnostic criteria for PF-ILD, non-survivors were characterized by significantly older age, a higher proportion of males, lower BMI, and a greater prevalence of reticulation and honeycombing observed on HRCT. They also exhibited elevated percentages of neutrophils and reduced percentages of macrophages and lymphocytes in bronchoalveolar lavage fluid (BALF), as well as higher levels of monocytes, neutrophils, and C-reactive protein (CRP) in serum. Pulmonary function was notably impaired in non-survivors, with significantly lower predicted values for FVC%pred, forced expiratory volume in 1 s (FEV1%pred), and DLCO%pred. In addition, the prevalence of comorbid conditions such as diabetes mellitus and gastroesophageal reflux disease (GERD) was significantly higher in non-survivors. The demographic characteristics of the CQ cohort are presented in S Tables 3 and 4.
Prognostic factors associated with mortality in PF-ILD: Results from univariate and multivariate analyses
Univariate and multivariate analyses were conducted to identify demographic, radiological, and clinical predictors of mortality in patients with PF-ILD based on the training cohorts, with the results summarized in S Table 5 (univariate analysis) and Table 2 (multivariate analysis). Continuous variables were categorized on the basis of cutoff values and previous literature11,21,24 (cutoff value of age = 65 years; cutoff value of FVC% pred = 97.2%; cutoff value of DLCO% pred = 47%; cutoff value of CRP = 12 mg/L). The multivariate analysis revealed that the ILD subgroups, baseline FVC%pred, baseline DLCO%pred, age at diagnosis, antifibrosis treatment, presence of GERD complication, CRP level, and BMI were significant independent predictors of mortality. Compared to patients with sarcoidosis, the risk of mortality was markedly higher in those diagnosed with IPF (HR 11.57, 95% CI (6.42, 20.85)), non-IPF IIP (HR 6.67, 95% CI (3.71, 12.00)), autoimmune-related ILD (HR 6.54, 95% CI (3.65, 11.70)), or exposure-related ILD (HR 4.21, 95% CI (2.19, 8.08)). Decline in pulmonary function was associated with increased mortality; patients with FVC% ⩾ 95% or DLCO% ⩾ 40% had significantly lower mortality risk than those with FVC% < 95% or DLCO% < 40%. Antifibrotic therapy was associated with a protective effect, as patients receiving such treatment had a reduced mortality risk (HR 1.59, 95% CI (1.34, 1.90)), compared to untreated patients. Advanced age (⩾65 years) was associated with a higher risk of death (HR 2.22, 95% CI (1.78, 2.75)), compared to those aged < 55 years. Furthermore, the presence of GERD significantly increased mortality risk (HR 1.45, 95% CI (1.23, 1.70)), patients with CRP > 12 mg/L had more than double the risk of death compared to those with CRP < 12 mg/L (HR 2.47, 95% CI (2.08, 2.94)), and BMI ⩾ 20 kg/m2 was significantly associated with improved overall survival (HR 0.54, 95% CI (0.45, 0.64)).
Factors associated with mortality in multivariate Cox regression analysis.
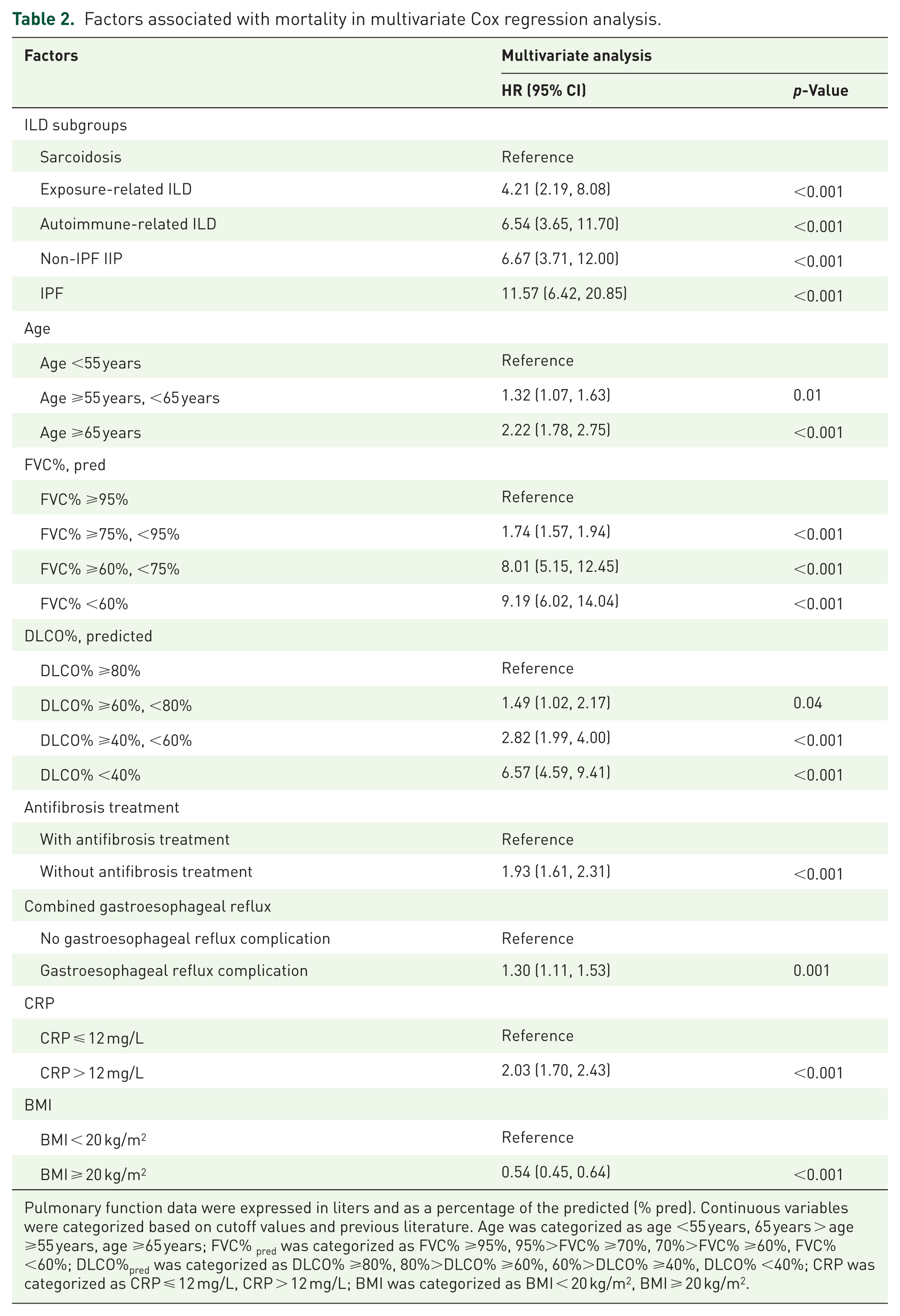
Pulmonary function data were expressed in liters and as a percentage of the predicted (% pred). Continuous variables were categorized based on cutoff values and previous literature. Age was categorized as age <55 years, 65 years > age ⩾55 years, age ⩾65 years; FVC% pred was categorized as FVC% ⩾95%, 95%>FVC% ⩾70%, 70%>FVC% ⩾60%, FVC% <60%; DLCO%pred was categorized as DLCO% ⩾80%, 80%>DLCO% ⩾60%, 60%>DLCO% ⩾40%, DLCO% <40%; CRP was categorized as CRP ⩽ 12 mg/L, CRP > 12 mg/L; BMI was categorized as BMI < 20 kg/m2, BMI ⩾ 20 kg/m2.
CI, confidence interval; CRP, C-reactive protein; DLCO, diffusion capacity of carbon monoxide; FVC, forced vital capacity; HR, hazard ratio; ILD, interstitial lung disease.
Establishment and validation of a nomogram-based mortality prediction model for PF-ILD
According to previous studies,8,26 we developed a mortality prediction model using the training set of PF-ILD patients from the BJ cohort. The model incorporated variables identified as statistically significant in the multivariate analysis, including ILD subtypes, baseline FVC%pred, baseline DLCO%pred, age at diagnosis, antifibrotic treatment, presence of GERD, CRP level, and BMI (Figure 1). Each risk factor was assigned a point value, and the cumulative score was used to estimate mortality risk. Notably, patients with a total score below 128.3 had a mortality rate of zero, whereas those with scores above 281.2 had a survival rate of zero.

A nomogram based on the multivariate Cox regression analysis and clinically meaningful parameters.
The calibration curve demonstrated that the nomogram had excellent predictive accuracy, with minimal deviation between predicted and observed outcomes in both the training and validation cohorts (S Figure 2, a-h, i-p). ROC analysis further validated the model’s performance. In the training cohort, the AUC was 0.7 for 12-, 24-, 36-, 48-, and 60-month overall survival (OS), 0.8 for 84- and 96-month OS (S Figure 3a). In the validation cohort, the AUCs were 0.7 for 12-, 24-, 36-, 48-, 60-, and 72-month OS, 0.8 for 84- and 96-month OS) (S Figure 3b). These findings indicate that the model provides robust discrimination between survivors and non-survivors in patients with PF-ILD.
Sensitivity analysis confirms independent survival benefit of antifibrotic therapy in PF-ILD
We repeated the Cox regression analysis after excluding patients who had received any concomitant therapies. In our cohort, 393 patients (27.7%) received concomitant treatments. Univariate Cox regression analysis demonstrated that antifibrotic therapy remained significantly associated with reduced mortality, both before (HR = 0.70, 95% CI: 0.58–0.85, p < 0.001) and after (HR = 0.63, 95% CI: 0.46–0.85, p < 0.001) exclusion of patients receiving concomitant therapies. Consistently, multivariate Cox regression analysis showed that antifibrotic therapy independently reduced the risk of death, with hazard ratios of 0.58 (95% CI: 0.48–0.71) and 0.52 (95% CI: 0.43–0.62), respectively, before and after excluding patients receiving concomitant therapies.
Survival analysis and prognostic impact of clinical factors in PF-ILD patients
The median OS time was 53 months in the BJ cohort and 43.5 months in the CQ cohort. By the last follow-up, 10.57% (n = 150) of patients in the BJ cohort had undergone lung transplantation, while 43.55% (n = 618) had died. In the CQ cohort, 4.26% (n = 12) received lung transplantation, and 42.55% (n = 120) died.
Kaplan–Meier survival curves stratified by the predictive model factors for both the BJ and CQ cohorts are shown in Figures 2 and S Figure 4. Among the ILD subgroups, exposure-related ILD and sarcoidosis did not reach a 50% mortality rate during follow-up. Patients with non-IPF IIP and autoimmune-related ILD had similar median survival times of 63 and 65 months, respectively, whereas IPF patients had a markedly shorter median survival of 30 months. The differences among ILD subgroups were statistically significant (p < 0.001).

Survival estimates performed by Kaplan–Meier curve. Survival estimates were performed using the Kaplan–Meier method for the (a) ILD subgroup, (b) baseline FVC%pred, (c) baseline DlCO%pred, (d) age at diagnosis, (e) antifibrosis treatment, (f) combined gastroesophageal reflux, (g) CRP level, (h) BMI in the training cohort. The overall baseline patient cohort was defined as the date of the disease progression. The patients were censored at the time of their last clinic visit or mortality.
PFT analysis revealed that patients with FVC%pred ⩾ 95% or DLCO%pred ⩾ 60% did not reach a 50% mortality rate during follow-up. In contrast, median survival times significantly decreased as lung function declined (p < 0.001). Specifically, patients with FVC%pred < 60% or DLCO%pred < 40% had a halved median survival.
Age was also significantly associated with mortality risk (p < 0.001). Patients aged over 65 years had a median survival time that was nearly half of the overall cohort. The use of antifibrotic therapy was associated with a survival benefit, extending median survival by 18 months compared with those not receiving such treatment (p < 0.001). Conversely, patients with GERD had a median survival that was 29 months shorter than those without GERD (p < 0.001). Elevated CRP levels (>12 mg/L) were also associated with a significantly worse prognosis, with median survival reduced by half compared to patients with CRP ⩽ 12 mg/L (p < 0.001). The median survival time for patients with a BMI ⩾ 20 kg/m² was 91 months, while for those with a BMI < 20 kg/m², it was 28 months (p < 0.001).
Distinct long-term pulmonary function trajectories among PF-ILD subtypes
Previous studies have demonstrated that a decline in FVC of more than 10% is the strongest predictor of reduced transplant-free survival (TFS), conferring more than a threefold increased risk of death or lung transplantation in both U.S. and UK cohorts. In our prior analysis, baseline FVC and DLCO were identified as independent predictors of survival. Building on these findings, a longitudinal assessment of FVC and DLCO in the BJ cohort following the diagnosis of PF-ILD revealed distinct functional trajectories between survival outcomes. Patients with PF-ILD who died exhibited a markedly greater decline in both FVC and DLCO over time compared with those who survived (Figure 3). While FVC remained relatively stable in the survival group during the first 3 years, a sharp decrease was observed in the death group beginning in year four, with progressive worsening through year six. DLCO followed a similar but earlier pattern of divergence, with the death group showing significant reduction from as early as the second year post-diagnosis. At year 5, the mean FVC change was −0.168 (SD: 0.349) in survivors and −0.528 (SD: 0.868) in the deceased group. By year 6, the FVC reduction had reached −0.28 (SD: 0.736) and −0.629 (SD: 0.774), respectively. Similarly, DLCO changes at year 5 were −0.26 (SD: 0.17) in the survival group and −1.56 (SD: 0.11) in the death group. The decline in DLCO persisted at year 6, with values of −0.78 (SD: 0.22) versus −1.37 (SD: 0.14), respectively. Due to the high proportion of missing PFT data at 84 and 96 months (both > 30%), these time points were excluded from the pulmonary function trend analysis to avoid potential bias. These findings suggest that dynamic declines in both FVC and DLCO, particularly after the third year, are associated with worse prognosis and may serve as valuable longitudinal biomarkers for risk stratification in PF-ILD.
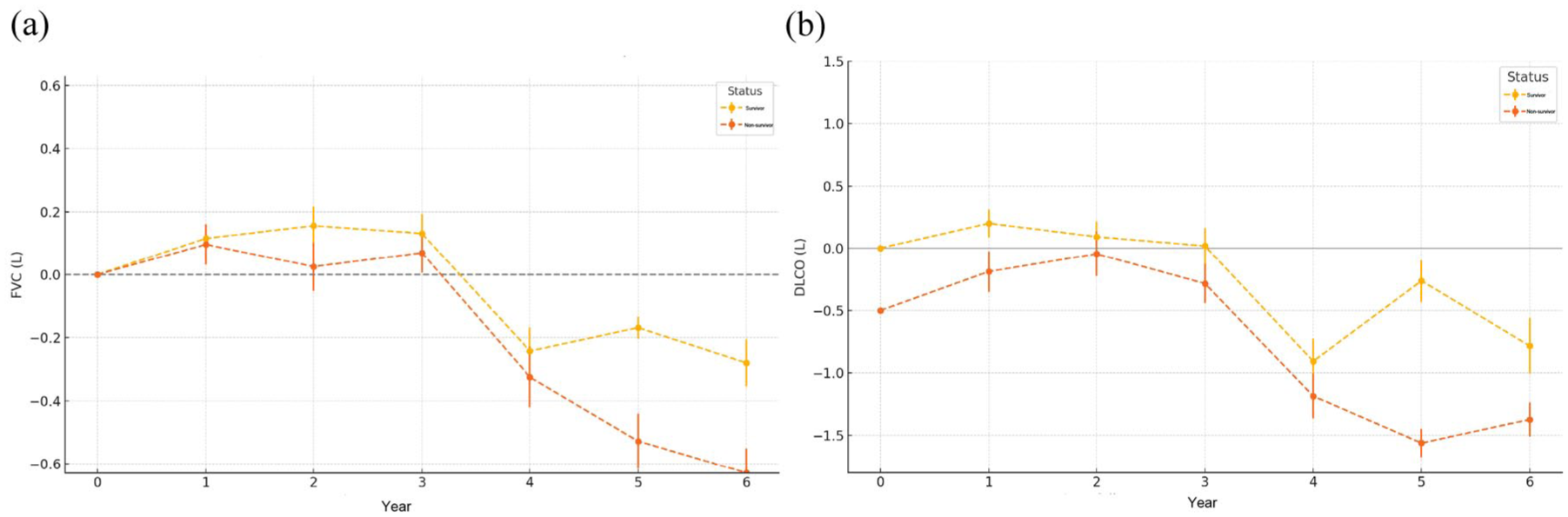
Annual changes in pulmonary function in patients with PF-ILD stratified by survival status. (a-b) Longitudinal trends in FVC (left) and DLCO (right) in patients with PF-ILD, comparing survivors (yellow) and non-survivors (red) over a 6-year period. Data are presented as mean ± standard error.
We further evaluated pulmonary function trajectories stratified by ILD subtype and found significant differences in both FVC and DLCO over time (S Figure 5). Longitudinal analysis revealed that changes in pulmonary function significantly diverged among subgroups (p < 0.001 for both FVC and DLCO). Patients with sarcoidosis, autoimmune-related ILD, or exposure-related ILD demonstrated a consistent upward trend in FVC from baseline, suggesting potential pulmonary recovery or stabilization over time. In contrast, individuals with IPF or non-IPF IIP exhibited a progressive and sustained decline in FVC, with IPF patients showing the most pronounced deterioration. Notably, the trend in DLCO closely paralleled that of FVC, further reinforcing the subgroup-specific differences in disease progression.
Baseline predictors and prognostic impact of AEs
Baseline characteristics were compared between patients who experienced AEs and those who did not in the training cohort (S Table 6). At the time of PF-ILD diagnosis, patients who subsequently developed AEs were significantly older, more likely to be male, had a lower BMI, and showed a higher prevalence of reticulation and honeycombing patterns on HRCT. Laboratory findings revealed that AE patients had elevated serum CRP levels, higher neutrophil percentages, and lower lymphocyte percentages in BALF, indicating a heightened inflammatory state. Pulmonary function was significantly more impaired in AE patients, with lower predicted values for FVC%, FEV1%, and DLCO%. Importantly, patients receiving antifibrotic therapy demonstrated a significantly lower risk of developing AEs. Moreover, during the 8-year follow-up period, patients who experienced AEs had markedly worse survival outcomes, with a median survival of 26 months compared with 109 months in patients without AEs (p < 0.001; S Figure 6).
Discussion
To precisely manage patients with PF-ILD, we constructed a clinical prediction model for 8-year survival with high specificity and sensitivity based on two distinct cohorts. This clinical prediction model integrated various associated factors, including ILD pathological subgroup types, baseline FVC%pred and DLCO%pred, age, antifibrosis treatment, GERD complication, CRP level, and BMI. This model may help quickly identify high-risk populations and guide necessary interventions in PF progression.
Surveillance of PF-ILD progression includes multiple factors, such as symptoms, serial lung function measurements, fibrotic extent measurement through HRCT, exercise capacity assessments, need for supportive care, and potential serum biomarkers.3,20,27 There is a paucity of studies focused on PF-ILD subtypes, although many studies on the prognostic factors of IPF,10,28 autoimmune-related ILD, 11 and another signal subgroup of ILD have been published. 24 Our study first used subtypes as one of the prognostic predictors and assessed the proposed mortality occurrence in patients with PF-ILD using a clinical prediction model with high specificity and sensitivity. This model can provide 8-year survival predictions based on clinical parameters and calculates the scores of each patient to predict the 8-year mortality risk.
The baseline demographic characteristics of patients with the PF-ILD phenotype in our cohort were generally consistent with those of the placebo arm of the INBUILD study, demonstrating that the clinical manifestations of PF-ILD have no distinction between our cohort and that in 15 American, European, and Asian countries. 4 The results of the ROGRESS study showed that older age, male sex, and ILD diagnosis are predictors of mortality in PF-ILD patients in France, 29 which coincided with our study that both advanced age and male sex may shorten patient survival time. Nasser indicated that hypersensitivity pneumonitis (HP) and sarcoidosis had the lowest mortality risk. 29 This was confirmed in the Chinese cohort used in this study. Previous studies have suggested that baseline lung function and decline in lung function are strong predictors of progressive fibrosis.21,26,30 In this study, baseline FVC%pred and DLCO%pred were strong predictors of mortality. However, there was no significant difference in the decline of lung function in patients with PF-ILD between survivors and non-survivors, which might be because the number of patients with PF-ILD with longitudinal lung function data was only 1/4 of the included cohort. To obtain an accurate conclusion, more complete follow-up PFT data are required for validation. Several studies have reported that higher CRP levels are associated with poorer outcomes in patients.31–33 The median survival of patients with CRP > 12 mg/L was half that of patients with CRP levels ⩽ 12 mg/L in our study. This may reflect the continuous pathogenic process in patients with PF-ILD, including damage to the alveolar epithelium and blood vessels, and activation of the inflammatory cytokine pathway. Weight loss has been previously reported to be associated with poor outcomes in various respiratory diseases34,35 and is closely linked to changes in pulmonary function.36,37 Consistently, our study demonstrated that a BMI of < 20 kg/m2 was associated with a significantly increased risk of mortality, nearly doubling the hazard of death compared with patients with a BMI ⩾ 20 kg/m2.
Regarding intervention methods, the INBUILD trial 4 and RELIEF study 6 suggested that in patients with PF-ILDs other than IPF who deteriorate using conventional therapy, combining antifibrotic drugs with existing treatment may attenuate disease progression, as measured by decline in FVC. However, no significant improvement in survival was observed in either trial. In the present study, the median survival of patients with PF-ILD, including IPF, treated with antifibrotic drugs, was 18 months longer than that of patients without antifibrotic drug treatment. This finding revealed a gap between clinical studies and real-world analyses of the effects of antifibrotic drugs. While concomitant therapies are indeed associated with the survival outcomes of patients with PF-ILD, they may potentially overestimate the impact of antifibrotic treatment. To address this, a sensitivity analysis was conducted, which demonstrated that antifibrotic therapy significantly reduced the risk of death both before and after excluding patients receiving concomitant therapies. This confirms that the survival benefit of antifibrotic treatment remains robust and is not confounded by the use of concomitant interventions. However, future prospective studies should consider excluding or stratifying patients based on concomitant therapy use to minimize bias and more accurately assess the effects of treatment. Alqalyoobi suggested that reflux/dysphagia symptoms resulting from esophageal motility dysfunction and chronic microaspiration were strong predictors of FVC decline over time 38 and could cause persistent alveolar epithelial damage, which might accelerate pulmonary fibrosis. We also observed that the median survival of patients with GERD was 29 months shorter than that of patients without GERD.
We combined the risk factors mentioned above into a predictive model and used it as a simple risk-screening method for patients with PF-ILD. Moreover, this model allows more precise risk assessment for each patient, potentially informing their management. We externally validated this model in a distinct cohort and found that it had good generalizability. In terms of clinical usability, the score is calculated based on routine clinical parameters that are typically available in the electronic medical record or easily obtainable during the diagnostic workup. Thus, we believe that the model is feasible to implement in day-to-day practice once a patient is confirmed to have met the criteria for PF-ILD. Exposure to antifibrotic treatment was defined as the regular use of antifibrotic agents for more than 3 months after ILD diagnosis and before meeting the PF-ILD criteria. Therefore, this model is intended for use at the time of PF-ILD diagnosis, not at the initial ILD diagnosis or the first clinical visit. By that time, the patient’s antifibrotic treatment status can be determined, allowing accurate input into the scoring system. To demonstrate the usefulness of this model as an aid in management decisions, we described its application in patients with PF-ILD. For example, in a 45-year-old male patient with autoimmune-related ILD, DLCO%pred was 65%, FVC%pred was 76%, treated with pirfenidone, without complicated GERD, with a CRP level of 8 mg/L, BMI > 20 kg/m2 and a model-calculated score of 51.12, corresponding to an 8-year risk of death of zero. These patients do not require a high level of clinical concern. We believe this model could provide a framework for clinicians and patients with PF-ILD to discuss prognosis and serve as a tool for policymakers to identify high-risk populations, thereby maximizing the efficiency and capacity of clinical trials.
We have previously confirmed that baseline FVC and DLCO are strong predictors of mortality in patients with interstitial lung disease (ILD). Near-term disease progression, particularly as measured by changes in FVC, is widely adopted as a primary endpoint in ILD clinical trials, given its well-established association with survival outcomes. In our longitudinal analysis, we found that FVC remained relatively stable during the first 3 years following the diagnosis of progressive fibrosing ILD (PF-ILD) in the survival group, while a marked and progressive decline began in the death group starting in year four and continued through year six. A similar trend was observed for DLCO, with a more rapid and earlier decline seen in non-survivors. These findings suggest that pulmonary function trends beyond the third year after PF-ILD diagnosis are closely associated with long-term prognosis and may serve as valuable indicators for risk stratification and disease monitoring.
When stratifying by ILD subtype, we found that FVC and DLCO declined to varying degrees in both IPF and non-IPF IIP, whereas they increased in sarcoidosis, autoimmune-associated ILD, and exposure-associated ILD. This might be due to a wide range of systemic manifestations in autoimmune-related ILD rather than most ILDs with predominantly pulmonary disorders, and the administration of immunosuppressive agents may attenuate these symptoms. 39 The PFT of sarcoidosis patients or exposure-related ILD with PF-ILD phenotype recorded in this study was in line with a previous study.13,40 Pulmonary sarcoidosis is a benign and self-limiting disorder that occurs in most patients. 41 Clinically occult disease and spontaneous remission occur in the majority of patients, and impaired lung function may improve with hormone therapy or on its own. 42 This may explain why we observed progressive improvements in FVC and DLCO in patients with sarcoidosis. Chronic hypersensitivity pneumonitis (CHP) is an immune-mediated ILD that results from an immune-mediated response in populations susceptible and sensitized to large amounts of inhaled antigens in the environment. 40 Clinical manifestations are influenced by factors including the nature and amount of inhaled antigens. The majority of HP patients recover after removal from the allergen, 43 which explains our observation in the present study that PFT in patients with HP tends to recover gradually. Our study has already pointed out that lower baseline FVC%pred and DLCO%pred are significantly associated with an increased risk of mortality. However, during the later follow-up period, we found that the changes in lung function in the different ILD subgroups of patients were not fully consistent with the risk of mortality. Patients with IIP experienced an annual decrease in FVC and DLCO, which was consistent with an increased mortality risk, whereas exposure-related ILD and autoimmune-associated ILD experienced an annual increase in FVC and DLCO during the follow-up, which was not in line with an increased mortality risk. The change in FVC after satisfying the proposed PF-ILD criteria is also heterogeneous depending on the criterion of interest and is strongly influenced by ILD diagnosis. Oldham et al. reported that changes in FVC after meeting the proposed PF-ILD criteria were strongly influenced by ILD diagnosis, 26 but they did not examine the association between survival rate and FVC changes within ILD subgroups. Pugashetti indicated that FVC decline strongly predicted reduced transplant-free survival (TFS), and in the absence of a 10% FVC decline, TFS was highly variable depending on the ILD subtype. 21 However, our study identified subgroups as a strong predictor of mortality, but due to the small sample size with follow-up lung function data, we did not find that a decline in FVC or DLCO was a risk factor for death in patients with PF-ILD, and whether subgroups have the same predictive power at different levels of change in lung function was not validated. Future studies with larger samples are needed to further explore this finding.
AEs (acute exacerbations) have been shown to be strongly associated with high mortality and may represent a critical point of irreversible decline in the disease trajectory. Previous studies have reported an annual incidence of AEs in IPF ranging from 5% to 9%, with in-hospital mortality reaching 50% to 80%.44,45 AEs have also been observed in patients with non-IPF ILDs, including PPF.46–48 The prognostic impact of AEs has been well documented in both IPF49–51 and non-IPF fibrosing ILDs.52,53 For example, Suzuki et al. studied 557 patients with non-IPF fibrosing ILD and found that those who experienced AEs had significantly worse outcomes compared to those who did not 47 (median survival: 4.3 years vs not reached; p < 0.001). These findings highlight the importance of close monitoring and timely intervention in patients with PF-ILD who develop AEs, as their prognosis may be markedly worse. In our research, we also observed that patients who experienced AEs during the 8-year follow-up had a survival rate approximately 1/3 shorter than those who did not experience AEs. Furthermore, independent risk factors associated with survival were found to be significantly linked to AEs, suggesting that the occurrence of AEs is closely related to poorer prognostic outcomes in PF-ILD patients. In future studies, the timing of AEs during the follow-up process should also be given careful attention, as it may further refine prognostic predictions
Conclusion
A clinical prediction model based on ILD subgroups, baseline FVC%pred, baseline DLCO%pred, age, antifibrosis drug usage, GERD complications, and CRP level could predict 8-year survival with high specificity. In addition to these baseline predictors, progressive declines in FVC and DLCO were strongly associated with poor prognosis and may serve as valuable longitudinal biomarkers for ongoing risk stratification.
Limitation
This study has several limitations that should be acknowledged. First, as a retrospective multicenter study, it is inherently subject to potential selection bias and missing data, which may affect the generalizability of the findings. Although strict inclusion criteria were applied, the accuracy of diagnostic classification may have been influenced by inter-center variability in clinical judgment. Second, several clinically relevant variables, such as patient-reported outcomes, environmental exposure history, and medication adherence, were not consistently available in the medical records and therefore could not be incorporated into the analysis. Third, the use of concomitant therapies may have confounded the observed effect of antifibrotic treatment. Although we performed a sensitivity analysis that confirmed the survival benefit of antifibrotic therapy remained significant after excluding patients receiving concomitant treatments, the possibility of residual confounding cannot be fully excluded. Future prospective studies should consider excluding or stratifying patients based on concomitant therapy use to minimize bias and more accurately assess treatment effects. Fourth, the study did not specifically account for neurological comorbidities, which may significantly impact overall mortality and functional outcomes. Future investigations should aim to include comprehensive assessments of neurologic conditions to better isolate the contribution of pulmonary fibrosis to survival outcomes. Besides, there was a high proportion of missing PFT data at 84 and 94 months (both > 30%). More complete long-term pulmonary function data should be collected in future research to accurately reflect the trajectory of functional change in patients with PF-ILD. Finally, as this study is a retrospective analysis utilizing the Cox regression model, no power calculation or sample size estimation was performed prior to data collection. This may limit the statistical power of the findings, and the results should be interpreted with caution. Future prospective studies with a calculated sample size would help validate these findings and provide more robust evidence.
Supplemental Material
sj-doc-7-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-doc-7-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-10-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-docx-10-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-11-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-docx-11-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-12-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-docx-12-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-8-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-docx-8-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-9-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-docx-9-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tif-1-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tif-1-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tif-2-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tif-2-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tif-3-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tif-3-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tif-4-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tif-4-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tif-6-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tif-6-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-tiff-5-tar-10.1177_17534666251379586 – Supplemental material for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients
Supplemental material, sj-tiff-5-tar-10.1177_17534666251379586 for A clinical predictive model for the long-term survival of progressive fibrosis interstitial lung disease patients by Jin-Min Gu, Si-Yao Xiao, Bo-Tao Chen, Shao-Ting Kang, Wei-Chao Li, Mei-Yi Zhang, Yang Yu and Gui-Ling Han in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.