
Abstract
Sarcoidosis is a multisystem disease with an unknown etiology and is characterized by the formation of noncaseating granulomas in the affected organs. We present the case of a 69-year-old male Japanese patient with bilateral hilar lymphadenopathy on chest radiographs for more than 10 years, left without further investigation. The patient reported no clinical symptoms. Chest computed tomography revealed ground-glass opacities and reticular shadows in both lungs, along with bilateral hilar and mediastinal lymphadenopathy. Lymphocytosis was observed in bronchoalveolar lavage fluid. Pathological examination of transbronchial lung biopsy revealed noncaseating, epithelioid granulomas congruous with sarcoidosis, together with other findings. There were no abnormalities on electrocardiogram, echocardiogram, and ophthalmic examination.
For progressive dyspnea on exertion, systemic corticosteroid therapy with oral prednisolone (25 mg/day) was initiated in 2017 and gradually tapered. Despite this intervention, the decline in forced vital capacity (FVC) was accelerated. Three years later, the patient noticed swelling in his right wrist. Further investigation revealed elevated anti-cyclic citrullinated peptide antibodies and absence of noncaseating epithelioid granuloma on surgical biopsy, leading to the diagnosis of rheumatoid arthritis (RA). Thereafter, the anti-fibrotic agent nintedanib was initiated, because interstitial lung disease (ILD) was considered to have converted into a progressive fibrosing phenotype (PF-ILD) with overlapping RA-associated lung involvement. With treatment, the progression of decline in FVC was slowed, although home oxygen therapy was introduced.
Keywords
Background
Sarcoidosis is a chronic systemic disorder of unknown etiology, and is characterized by the formation of noncaseating epithelioid granulomas in multiple tissues and organs. 1 It is difficult to predict the clinical course of sarcoidosis because it has a broad spectrum of clinical phenotypes ranging from acute to chronic; some patients may also be asymptomatic with spontaneous remission.2,3 Lung involvement is the most common presentation, characterized by bilateral hilar and mediastinal lymphadenopathy and reticular opacities on chest computed tomography (CT), 4 followed by skin, 5 eyes, 6 and joints. 7 Some patients with serious organ involvement require potent immunosuppressive treatments including corticosteroids, methotrexate, azathioprine, and anti-tumor necrosis factor-α (TNF-α) agents.8–10 Rheumatological manifestations can be the initial symptom of the disease. 11
Rheumatoid arthritis (RA) is a chronic autoimmune disorder of the joints, characterized by erosive arthritis and extra-articular involvement. 12 Sarcoidosis complicated by RA has only been sporadically reported. Overall, the clinical presentations differ between sarcoidosis and RA; however, they are both immune-mediated diseases that can lead to interstitial lung disease (ILD) as one of their most serious manifestations.13,14
Progressive fibrosing interstitial lung disease (PF-ILD), recently recognized as a phenotype of ILD, shares a disease behavior similar to that of idiopathic pulmonary fibrosis (IPF).15,16 PF-ILD is experienced in 20% of patients with sarcoidosis and approximately 25% of RA-associated ILD patients. 17 Although treatment of PF-ILD that develops under these conditions has yet to be established, a phase III clinical trial (INBUILD trial) 18 showed a favorable efficacy profile of nintedanib, a low molecular weight, triple-tyrosine kinase inhibitor that exhibits anti-fibrotic properties by blocking the signaling of platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF). 19 In the INBUILD trial, nintedanib significantly reduced the annual decline in forced vital capacity (FVC) in patients with non-IPF PF-ILD, including sarcoidosis and RA-associated ILD, leading to its approval for treatment of PF-ILD in several countries including the United States, Canada, and Japan.
Herein, we describe a rare case of sarcoidosis accompanied by RA, in which PF-ILD required treatment with nintedanib due to progressive respiratory failure.
Case report
A 68-year-old Japanese male patient was previously referred to a hospital in November 2012 for further investigation of bilateral hilar lymphadenopathy detected on a chest radiograph, which had been brought to notice at every annual medical checkup for the past 10 years but had not been examined further. The patient had no health complaints or particular medical history. He had worked in sales and had a 15 pack-year smoking history. Chest radiography revealed bilateral hilar lymphadenopathy and reticular shadows in the lower lung fields (Figure 1(a)). CT of the chest showed bilateral hilar and mediastinal lymphadenopathy (Figure 1(b)), and ground-glass opacities and reticular shadows distributed predominantly in both lower lobes (Figure 1(c)).
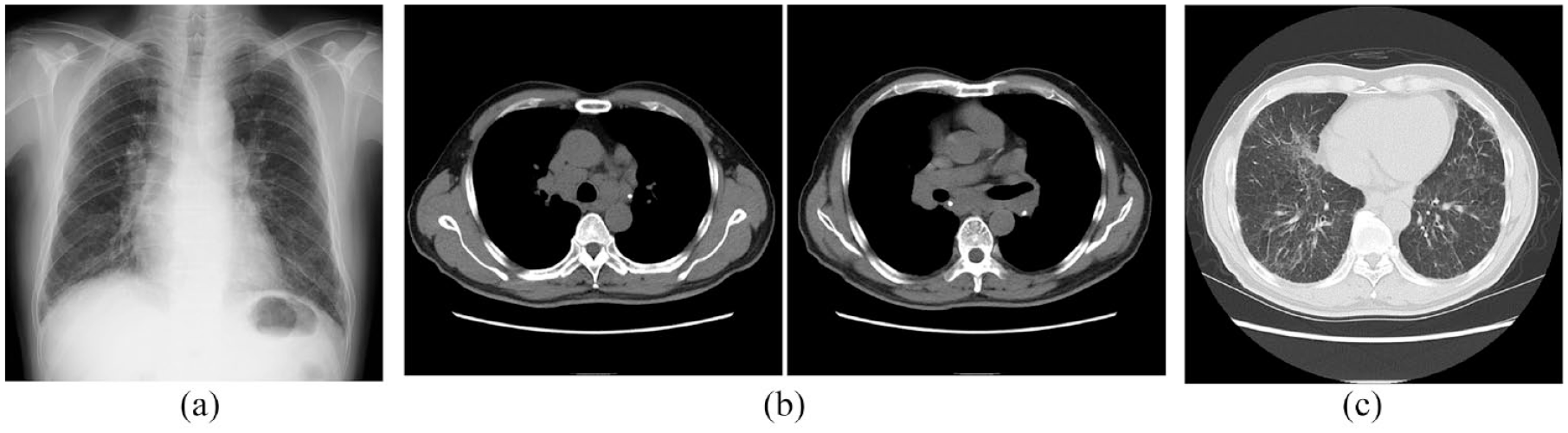
(a) Chest X-ray taken in November 2012 during the patient’s first hospital visit shows stage II (bilateral hilar lymphadenopathy with pulmonary infiltration) disease in chest X-ray staging. (b) Computed tomography (CT) of the chest shows bilateral hilar and mediastinal lymphadenopathy. (c) The CT scan also shows ground-glass opacities and reticular shadows distributed predominantly in both lower lobes.
The vital signs were as follows: body temperature, 36.3°C; blood pressure, 142/72 mmHg; pulse, regular at 72 beats/min; oxygen saturation, 97% (ambient air). Physical examination revealed no abnormalities except for fine crackles in the bilateral lower lung fields. An ophthalmic examination showed no findings of granulomatous uveitis.
Laboratory investigations showed that the C-reactive protein (CRP) level was 0.09 mg/dL (normal < 0.3 mg/dL). Increased levels of lactate dehydrogenase (LDH) (233 U/L, normal < 211 U/L) and Krebs von den Lungen-6 (KL-6) (1121 U/mL, normal < 500 U/mL) were observed. Serological tests showed an anti-cyclic citrullinated peptide (CCP) antibody level of <1.2 U/mL (normal < 4.5 U/mL) and rheumatoid factor (RF) of <1.0 IU/mL (normal < 10.0 IU/mL). The patient’s serum angiotensin-converting enzyme (ACE) level was 24.6 U/L (normal < 25.0 U/L).
Pulmonary function test (PFT) revealed mild reduction in FVC, 2.96 L (87.3% predicted), forced expiratory volume in 1 s (FEV1.0), 2.38 L (91.9% predicted), and diffusion capacity of the lung for carbon monoxide (DLCO), 14.03 mL/min/mmHg (83.4% predicted). Although electrocardiography showed complete right bundle branch block, the transthoracic echocardiogram revealed no abnormality.
Bronchoscopy revealed no macroscopic abnormalities. Bronchoalveolar lavage fluid (BALF) analysis showed a cell count of 2.43 × 105/mL, with a cell differentiation of 39.0% macrophages, 61.0% lymphocytes, and a CD4/CD8 ratio of 1.9. Histopathological examination of the transbronchial lung biopsy (TBLB) specimens (right B3b, right B8a) revealed noncaseating epithelioid granulomas in the perivascular connective tissue with lymphocyte infiltration, which was consistent with sarcoidosis (Figure 2(a) and (b)). Alcian blue-positive intraluminal fibrosis with mural incorporation was also observed (Figure 2(c)). No microorganisms, including Mycobacterium spp. and Propionibacterium acnes, were detected in BALF, tissue smears, or lung specimen cultures.

(a) A biopsy specimen obtained from the right B8a and subjected to histopathological analysis shows noncaseating epithelioid granulomas (arrows, ×100). (b) A granuloma specimen under ×400 magnification. (c) Magnified pictures (×400) of a rectangle within the left upper panel are shown. Alcian blue positive intraluminal fibrosis with mural incorporation is observed (left lower panel).
In May 2016, the patient was referred to our hospital. Due to a progressive decline in FVC (>200 mL/year) accompanied by a deterioration of the radiological findings, 25 mg/day oral prednisolone (PSL) was initiated in November 2017 followed by gradual tapering (Figure 3). In March 2020, when PSL was reduced to 12 mg/day, the patient noticed arthralgia and swelling in the right wrist followed by the right third PIP and the left wrist, but not in ankle joints. No skin lesions were found. Although a giant cell tumor of the tendon sheath was tentatively suspected, histopathological examination of the joint lesion showed proliferative synovial tissues with massive infiltration of mononuclear cells, but not granuloma formation or giant cell tumor. Mgnetic resonance imagine (MRI) revealed bone marrow edema in the carpal bones and synovitis in the right wrist. In addition to these findings, a high anti-CCP antibody titer (>500 U/mL) led to the diagnosis of RA in August 2020, though the clinical joint findings were not necessarily typical. Salazosulfapyridine was initiated for the joint lesions.

A summary of the patient’s clinical course.
Gradual progression of dyspnea had been accompanied by a total of 900 mL reduction in FVC during the 4 years until August 2020 (Figure 3). Concordantly, honeycomb changes gradually developed on a series of CT scans over 5 years (Figure 4). Nintedanib was initiated for PF-ILD in January 2021, resulting in a slowing of the FVC decline without the incidence of adverse events such as diarrhea or liver injury. Although home oxygen therapy was introduced in May 2021, FVC was maintained in follow-up pulmonary function tests with successful tapering of PSL to 10 mg/day (Figure 3). As of August 2021, the patient had stable symptoms and chest CT findings and was on regular monthly follow-up.

Honeycomb changes gradually developed on a series of computed tomography scans over 5 years of observation.
Discussion
In this report, we describe a case of concurrent sarcoidosis and RA in which the progression of ILD had accelerated for 4 years prior to the diagnosis of RA but was successfully controlled under treatment with nintedanib. Both sarcoidosis and RA are immune-mediated diseases of unknown etiology, though the role of genetic and environmental factors has been speculated. Kucera et al. 20 have shown HLA-DR4 as a common risk factor for co-occurrence of these two conditions, suggesting genetic predisposition for the overlapping phenomenon. However, unlike that in the present case, RA preceded sarcoidosis in all patients described in previous case reports,20–25 except that in the case report by Yutani et al. 26
Intriguingly, there have been many reported cases in which patients with RA developed sarcoidosis after anti-TNF-α administration.27,28 Anti-TNF-α treatment-induced sarcoidosis and/or sarcoid-like reactions have also been reported in patients with psoriatic arthritis, 29 ankylosing spondylitis, 30 and inflammatory bowel diseases, 31 suggesting that the development of sarcoidosis is more likely to be associated with anti-TNF-α treatment than be due to the underlying disease. Indeed, in most cases, patients improve upon discontinuation of anti-TNF-α agents, with or without systemic corticosteroid use. 32 In the current patient, however, sarcoidosis developed prior to RA independent of anti-TNF-α treatment.
Recently, the lung has been speculated to be the initiating site of injury in anti-citrullinated peptide antibody (ACPA)-positive RA. 33 It was reported that the increased expression of citrullinated peptides can be detected by immunohistochemical analysis of lung tissue in RA-ILDs. 34 Moreover, among ACPA-positive RA patients, the ACPA levels were relatively higher in the BAL fluid than in the sera, suggesting that ACPAs are locally produced in the lungs of these patients. 35 Thus, the lung should be focused upon as a potential primary initiating site for ACPA-positive RA. Regarding the pulmonary origin of systemic inflammatory disorders, silica exposure increases the risk of both sarcoidosis and seropositive RA, suggesting a common pathogenic pathway underlying both diseases. 36 However, taking into account our patient’s occupational history, sarcoidosis-associated lung involvement may be implicated in the development of RA since pre-existing lesions in the lungs, where RA-related anti-CCP immune responses are considered to be initiated, often precede manifestations in the joints. 37
Arthritis is one of the cardinal manifestations in Löfgren’s syndrome (LS), a distinct form of sarcoidosis with low prevalence among Asian patients. 38 LS is characterized by fever, bilateral hilar lymphadenopathy, and erythema nodosum, in addition to bilateral ankle arthritis or periarticular inflammation. 39 Joint findings in this case were not typical of either LS or RA, though preceding long-term corticosteroid therapy for sarcoidosis might have modulated the clinical course of joint manifestations. Taken together, RA was considered more likely based on positive ACPA and histological examinations that did not show noncaseating epithelioid granuloma formation characteristic of sarcoidosis.
In this case, the initially observed ground-glass opacities developed into honeycomb-like structures over approximately 8 years, in concordance with the rapid annual decline in FVC comparable to the features of IPF. Akira et al. 40 reported that 20% of sarcoidosis patients without honeycomb-like pulmonary patterns at their first examination eventually developed them from ground-glass opacities, along with a decline in FVC, consistent with the changes observed in our patient. However, when honeycomb-like patterns are discerned on chest CT in cases of sarcoidosis, it can be difficult to assess whether they correspond to IPF, collagen disease-associated ILDs complicating sarcoidosis, or pulmonary sarcoidosis itself.41,42 Referring to the mechanism of honeycomb-like pattern development in pulmonary sarcoidosis, Sawahata et al. 42 implicated the distal concentration of traction bronchiectasis, which develops through shrinkage of consolidations. Although histological examination was not performed after the development of honeycomb-like changes, it is likely that a similar mechanism was involved in our patient. However, the pathological finding of TBLB, including intraluminal fibrosis with mural incorporation, was suggestive of the simultaneous progression of RA-associated ILD. Although the prognosis of RA-usual interstitial pneumonia (UIP) is not as unfavorable as that in IPF in general, 43 the current UIP pattern accompanied by rapid FVC decline could be attributable to the synergistic effects of the co-occurrence of sarcoidosis and RA.
For our patient, nintedanib was initiated for PF-ILD. Nintedanib has been approved for IPF as it reduces the annual rate of decline in FVC by approximately 50%, that is, from roughly 200 mL/year to 100 mL/year. 44 In the INBUILD trial, patients were required to meet at least one of the following criteria to be defined as having ILD progression within 24 months of screening, despite standard treatment with agents other than nintedanib or pirfenidone: (1) a relative decline in the FVC of at least 10% of the predicted value, a relative decline in the FVC of 5% to <10% of the predicted value, together with either (2) worsening of respiratory symptoms or (3) an increased extent of fibrosis on high-resolution CT, or (4) worsening of respiratory symptoms and an increased extent of fibrosis. Our patient refused to undergo PFT for two years before the initiation of nintedanib because the tremendous effort required to perform the test would make him feel exhausted. Therefore, we concluded that the patient’s ILD exhibited a progressive phenotype based on his symptoms and radiological findings.
To our knowledge, this is the first report describing PF-ILD in a patient with sarcoidosis complicated by RA, who required the administration of nintedanib and subsequently experienced a slowing in FVC decline. Further accumulation of similar cases is indispensable to evaluate the precise effects of anti-fibrotic therapy under these conditions.