
Abstract
Pulmonary hypertensive diseases have been classified by the World Health Organization (WHO) into five groups based on the pathophysiology and characteristics of each disease state. Several targeted therapeutic agents have been developed that combat the vascular remodeling in WHO Group 1 pulmonary arterial hypertension, however, the search for treatment solutions in other WHO Groups has been less fruitful. In this review, we focus on therapeutic options for patients with pulmonary hypertension and interstitial lung disease (PH-ILD). Investigations of targeted WHO Group 1 PAH therapies have largely failed to improve functional capacity, hemodynamics, oxygenation, quality of life, or survival in PH-ILD. In contrast, inhaled treprostinil was shown effective in the INCREASE Trial, a placebo-controlled study in which patients with PH-ILD treated with inhaled treprostinil demonstrated a 31-meter placebo-corrected improvement in the primary endpoint, 6-minute walk distance. Treatment with inhaled treprostinil was also associated with improvement in time to clinical worsening, fewer exacerbations of underlying lung disease, decrease in N-terminal pro-B-type natriuretic protein (NT-proBNP) levels, and improvement in forced vital capacity compared to placebo. In this review, we also elaborate on the current understanding of the pathobiology leading to PH-ILD with emphasis on the role of newer signaling pathways and mediators of vascular biology that may expand treatment options. Strategy and innovations for early detection and diagnosis are highlighted while underscoring the importance of early detection and diagnosis. A holistic and collaborative approach to the treatment of PH-ILD is outlined including a variety of adjunctive measures and the consideration of patient-reported outcome data in order to improve disease management outcomes.
Introduction
Pulmonary hypertension is a syndrome resulting from increased vascular resistance in the pulmonary circulation that can ultimately lead to right ventricular failure and death. The World Health Organization (WHO) has classified pulmonary hypertensive diseases into five groups based on pathophysiologic distinctions resulting in elevated pulmonary vascular resistance. Over the past two decades, there has been considerable progress in understanding the pathophysiology of pulmonary arterial hypertension (WHO Group 1). Several targeted therapies have been approved by governmental drug safety authorities for the treatment of WHO Group 1 pulmonary arterial hypertension (PAH). These therapies have resulted from our current level of understanding of abnormal pulmonary vascular endothelial function characterized by excessive production of endothelin, deficient levels of nitric oxide and prostacyclin production, and abnormal regulation of cellular proliferative forces. The imbalance in endogenous mediators of vasomotor tone and vascular cell proliferation leads to characteristic remodeling of the pulmonary vasculature in patients with PAH. While mechanisms known to affect vascular physiology and cell populations in PAH may also have roles in PH-ILD, this disease state is more complex and involves both vascular and parenchymal pathophysiology. It remains unclear if these two features of PH-ILD share pathophysiologic pathways. Survival studies have demonstrated a significant survival disadvantage in patients with pulmonary hypertension associated with interstitial lung disease (PH-ILD) compared to those in other WHO groups. The antifibrotics, pirfenidone, and nintedanib, have been shown to limit the progression of fibrosis in patients with interstitial lung disease, but attempts to identify an effective targeted therapy to impact pulmonary vascular disease in these patients have been unsuccessful until recently. In 2021, Waxman and colleagues presented the results from a landmark investigation (INCREASE Trial) demonstrating distinct benefits of inhaled treprostinil in patients with PH-ILD. Herein we review state-of-the-art therapeutic options for PH-ILD, summarize evidence pertaining to new signaling pathways that may broaden treatment options, discuss strategy and innovations for detection and diagnosis, and highlight the importance of collaborative care and adjunctive measures in order to achieve better outcomes and quality of life.
Epidemiology and survival
The fibrotic interstitial lung diseases (ILD) represent a heterogeneous array of over 200 fibrotic lung disorders, each with the potential to be complicated by pulmonary hypertension. From a global perspective, the incidence of ILD has been reported from 1 to 31.5 per 100,000 person-years with prevalence ranging from 6.3 to 71 per 100,000 persons. 1 The global prevalence of pulmonary hypertension in patients with ILD also varies greatly with reports ranging from 3% to 86%.2,3 These wide ranges reflect the predominance of ILD subtypes in different study populations, differences in disease stage, and methods used to diagnose pulmonary hypertension. Kaul and colleagues have reported a higher prevalence of idiopathic pulmonary fibrosis and sarcoidosis in North America and Europe, higher rates of hypersensitivity pneumonitis in Asia, and wide global variability in ILD associated with connective tissue disease. 1 The stage of ILD appears to affect pulmonary hypertension prevalence with 8%–15% IPF patients reported to have pulmonary hypertension at the time of initial evaluation, 30%–50% in more advanced disease, and over 60% in end-stage disease. 4 In a longitudinal study of patients with ILD referred for lung transplant, Nathan et al. found pulmonary hypertension in 39% at the time of admission to a waiting list and 86% roughly 8 months later at the time of transplant. 5
The accuracy of prevalence rates may depend on diagnostic methods. While pulmonary artery dimensions on imaging studies and estimates of pulmonary artery pressure by echocardiography are useful screening tools, right heart catheterization provides the only accurate assessment of pulmonary hemodynamics in patients with parenchymal lung disease. Right heart catheterization is often delayed in ILD patients until referral for transplant evaluation. Hemodynamic criteria clearly affect prevalence reporting with higher rates of PH detected at lower mean pulmonary artery pressure (mPAP) thresholds.6 –8 Most prevalence studies have relied on older hemodynamic criteria that preceded the most recent hemodynamic definition of pre-capillary pulmonary hypertension including a mPAP > 20 mmHg and pulmonary vascular resistance > 2 Wood units. 9
Patients with WHO Group 3 PH have a significant survival disadvantage compared to patients with pulmonary hypertension in other WHO groups and further, WHO Group 3 patients with IPF have the least favorable survival prognosis of all WHO Group 3 subtypes. 10 Nadrous and colleagues reported survival at 0.7 years in IPF patients with systolic pulmonary artery pressure (sPAP) > 50 mmHg on transthoracic echocardiogram compared to 4.8 years if the sPAP was <35 mmHg. 11 Similarly, when IPF patients were stratified according to sPAP by echocardiogram 1-year mortality was 19.9% for sPAP < 40 mmHg, 47% if sPAP was 40–59 mmHg, and 76% with sPAP > 60 mmHg. 12 These survival statistics underscore the importance of early detection of PH in patients with ILD in order to effect earlier treatment intervention.
Vascular pathobiology in PH-ILD
While WHO Group 3 pulmonary hypertension due to lung disease with hypoxia has generally been considered a consequence of hypoxic vasoconstriction, other mechanisms have been proposed to explain the development of PH in patients with ILD (Figure 1). Hypoxic vasoconstriction is an adaptive response to maintain systemic blood oxygen levels by directing pulmonary blood flow to functioning regions of the lung parenchyma and away from areas with relative hypoxia. With parenchymal destruction, loss of small vessels and capillaries can also lead to increased vascular resistance. However, the severity of pulmonary hypertension in ILD does not necessarily correlate with the degree of lung function impairment or radiographic damage. 13 The detailed relationship between vascular pathobiology and parenchymal fibrosis in PH-ILD remains to be defined and is a task complicated by the multiple phenotypes representing interstitial lung diseases.
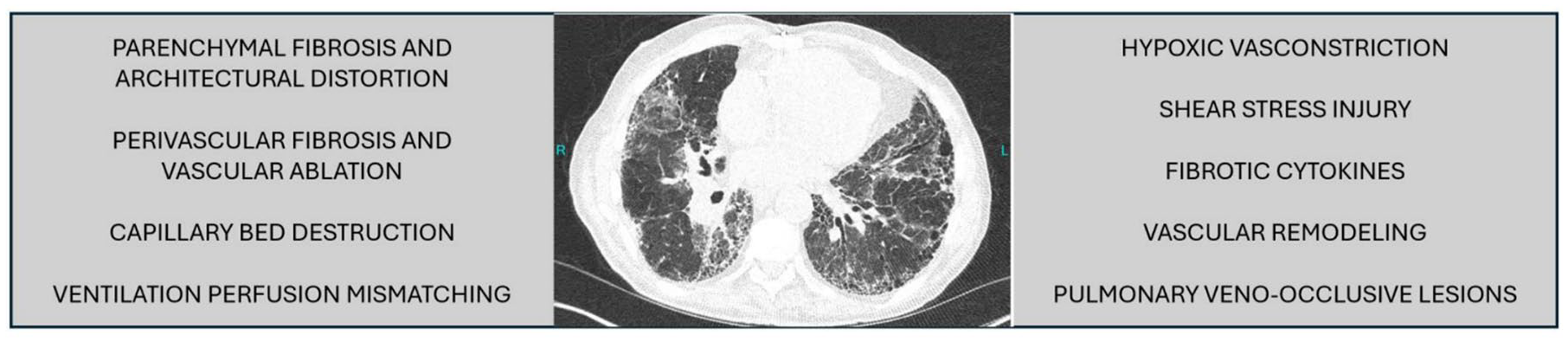
Possible mechanisms involved in the development of pulmonary hypertension in patients with ILD. 14
Contemporary investigations of PH-ILD are focused on the role of genetic, molecular, and intracellular mechanisms that drive cell proliferation, apoptosis, and inflammatory processes in both disease states. The discovery of the bone morphogenetic protein receptor-2 (BMPR-2) mutation associated with familial and some idiopathic cases of PAH sharpened the focus on intracellular signaling pathways of the transforming growth factor-beta (TGF-β) gene superfamily. Cytokines of the TGF-β superfamily affect multiple functions including cellular proliferation, differentiation, apoptosis, and inflammatory responses. BMPR-2 is a member of the TGF-β superfamily and serves as a counterbalancing force acting to suppress TGF-β signals that drive endothelial apoptosis and vascular smooth muscle proliferation. 14 The balance of BMPR-2/TGF-β signaling has been linked to the development of PH-ILD. In patients diagnosed with idiopathic pulmonary fibrosis (IPF), there is an inverse correlation between mean pulmonary artery pressure (mPAP) and BMPR-2 levels. 15 Reduced expression of BMPR-2 in myofibroblasts and pulmonary arterioles and increased expression of IL-6 and phosphorylated STAT3 (p-STAT3) have been observed in fibrotic lung explants from patients with IPF and PH compared to those with only IPF. 16 These findings were also associated with increased microRNAs which downregulate BMPR-2. Blocking IL-6 and p-STAT3 reduced microRNA expression and resulted in attenuation of fibrosis and vasculopathy.
Elevated endothelial production of endothelin 1 (ET-1) has long been known to play a key role in the vasculopathy of PAH and has also been observed in patients with IPF, especially when they have associated PH. 15 The endothelin type A receptor has been implicated in the promotion of TGF-B1-mediated fibrogenesis and the transition of epithelial to mesenchymal cells. 15 In a transgenic mouse model genetic overexpression of ET-1 has been linked to chronic lung inflammation and fibrosis with excessive accumulation of perivascular and peribronchiolar extracellular membrane proteins and CD4-positive mononuclear cells. 17
Chronic hypoxia in patients with ILD has been linked to several mediators of cell function that influence changes in vascular structure and biology similar to those noted in the vascular remodeling of PAH. Hypoxia is reported to increase ET-1 release and decrease nitric oxide (NO) production by endothelial cells, thereby disrupting normal vasomotor regulation.18,19 At the intracellular level, the pulmonary vascular response to hypoxia is known to be influenced by TGF-β1 and hypoxia-inducible factor 1-alpha (HIF-1α) signaling pathways.20 –22 HIF-1α is a subunit of the transcription factor hypoxia-inducible factor 1 (HIF-1) encoded by the HIF1A gene. 23 HIF-1 induces transcription of genes responsible for modulating angiogenesis, proliferation, and cell survival in response to changes in systemic oxygen level. 24 In experimental models, chronic hypoxia induces expression of vascular endothelial growth factor A (VEGFA) and its receptor VEGFR2 via HIF-1α resulting in endothelial proliferation.21,25 Endothelial-mesenchymal transition can be induced by TGF-β and HIF-2α under hypoxic conditions. 26 Hypoxia also leads to enhanced production of platelet-derived growth factor-beta (PDGF-B) which in turn promotes VEGFA-mediated endothelial proliferation.27,28 Serotonin, or 5-hydroxytryptamine (5-HT), has been implicated as a potential mediator linking hypoxia to the development of pulmonary hypertension via the overexpression of the 5-hydroxytryptamine transporter (5-HTT). 29 Hypoxia-induced overexpression of 5-HTT has been shown to promote 5-HT mitogenic activity in vascular smooth muscle, an effect that was blunted in 5-HTT deficient mice exposed to hypoxia.30,31 Although poorly defined overall, hypoxia appears to be a central factor driving multiple intersecting signals and mediators in PH-ILD similar to those causing vasculopathy in PAH.
Other mechanisms contributing to vasculopathy in patients with ILD may depend on genetic and molecular abnormalities that are not dependent on hypoxia or vessel destruction. A comparison of gene expression in human lung tissue from ILD patients with or without PH revealed a pro-inflammatory gene signature in patients without PH and a pro-proliferative signature if PH was present. 32 Increased vessel wall thickness can be found in both areas of fibrotic and non-fibrotic lung in patients with ILD and PH compared to those having ILD alone. 33 A potential link between specific phenotype and gene mutation has been suggested in a group of patients with PAH, low diffusing capacity, mild ILD and mutation of the VEGF receptor, kinase insert domain receptor (KDR). 34 One other mutation worth noting is mutation of the gene for T-box transcription factor 4 (TBX4) typically associated with the development of small patella syndrome, congenital heart disease, and cognitive impairment. PAH may occur in these patients with incomplete penetrance and may be associated with interstitial abnormalities such as septal thickening, micronodules and ground-glass opacities. 35
Experience with targeted PAH therapies in PH-ILD
Agents targeting pulmonary vascular pathobiology characteristic of WHO Group 1 PAH have yielded significant benefits in functional capacity, quality of life, time to clinical worsening, and extended survival in WHO Group 1 patients. Investigation of these agents in patients with WHO Group 3 PH associated with ILD has met with mixed results. The use of systemically administered vasoactive agents has the potential to disrupt ventilation–perfusion relationships and worsen oxygenation in the context of heterogeneous parenchymal disease. Furthermore, fibrotic lung disease may affect both pre- and post-capillary vessels leading to a response to systemic vasodilator therapy that mimics pulmonary veno-occlusive disease.
Investigations with endothelin receptor antagonists have failed to demonstrate meaningful benefits for patients with PH associated with ILD (Table 1). Bosentan use for pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia was examined in the BPHIT study, a randomized, placebo-controlled trial including patients with fibrotic idiopathic interstitial pneumonia (IIP) and PH confirmed by right heart catheterization. There was no improvement in the primary endpoint, pulmonary vascular resistance index (PVR index), symptoms, or functional capacity over the 16-week study period. 36 A lack of benefit with ambrisentan in patients with IPF and PH was also noted in the ARTEMIS-IPF trial which had been primarily designed to evaluate safety and efficacy of ambrisentan as a treatment for IPF. 37 ARTEMIS-IPF was discontinued early due to an increased risk of disease progression and hospitalization for respiratory deterioration. Subgroup analysis revealed similar findings in about 10% of subjects who also had PH. The subgroup findings, in turn, led to the early discontinuation of a companion study focused on patients with IPF and PH (ARTEMIS-PH).
Investigation of approved oral PAH therapies in PH-ILD.

6MW, 6-minute walk; BNP, brain natriuretic peptide; CO, cardiac output; IIP, idiopathic interstitial pneumonia; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; PAH, pulmonary arterial hypertension; PFT, pulmonary function test; PH, pulmonary hypertension; PH-ILD, pulmonary hypertension and interstitial lung disease; PVR, pulmonary vascular resistance; QOL, quality of life; RCT, randomized clinical trial; RHC, right heart catheterization; RVSD, right ventricular systolic dysfunction; RVSP, right ventricular systolic pressure; TID, three times daily.
Treatment of PH-ILD with nitric oxide (NO) pathway agents has met with mixed results (Table 1). A number of small studies of sildenafil without placebo comparison have suggested improvements in 6-minute walk (6MW) distance and brain natriuretic peptide (BNP) levels in patients with IPF and fibrotic ILDs with PH.38–43,45 Preservation of exercise capacity and improved quality of life were reported in a placebo-controlled study of sildenafil in a sample of patients with IPF and impaired right ventricular function. 46 The STEP-IPF trial evaluated sildenafil in patients with IPF and a DLCO < 35% predicted. Post hoc analysis revealed that patients with impaired RV function had a 99-m placebo-corrected improvement in 6MW distance. 47 The efficacy and safety of riociguat were studied in a range of ILD phenotypes with PH in the RISE-IIP trial. 44 There was no improvement in the primary endpoint of 6MW, and the study was discontinued early due to an inordinate number of deaths in the treatment arm, especially when patients transitioned from the 16-week placebo arm into the extension arm with riociguat. The 2022 ESC/ERS Guidelines suggest that phosphodiesterase-5 (PDE-5) inhibitors might be considered on a case-by-case basis for patients with ILD and PH depending on suspected severity of the vascular disease component. 9
Systemic prostanoid therapy has been used in patients with pulmonary hypertension and fibrotic lung disease with some reports suggesting at least short-term benefit. In one small retrospective review of 19 patients with pulmonary hypertension and moderate or severe ILD treated with epoprostenol (n = 10) or bosentan (n = 9), improvements in 6MW and functional class were noted initially, although significant deterioration was seen in nearly half at 1-year follow-up. 48 Systemic treprostinil therapy was associated with improvements in hemodynamics, echocardiographic function, 6-minute walk distance, and biomarkers without compromising systemic oxygenation in a small group of patients with pulmonary fibrosis and advanced PH referred for lung transplantation 49
Inhaled vasoactive therapy in PH-ILD
Inhaled agents are thought to be less likely to disrupt ventilation–perfusion relationships in areas of the lung with permanently impaired gas exchange and provide more favorable vasoactive effects without compromising systemic oxygenation. A comparison of intravenous epoprostenol, calcium blockade, inhaled NO and inhaled prostaglandin I2 in a small number of patients with lung fibrosis and PH addressed this issue and demonstrated preferential pulmonary vasodilation and decreased right to left shunting with inhaled NO and inhaled prostaglandin I2. Meanwhile, the systemic vasodilators did not discriminate between healthy and fibrotic lung, increased shunt flow, and also caused decreased systemic blood pressure. 50 One patient from this study was immobilized due to right heart failure and treated long term with inhaled iloprost. The patient experienced improved activity tolerance, a tendency toward improved hemodynamics, and was able to complete a 6-minute walk distance of 314 m after inhalation of iloprost at 12 months. In contrast, a 12-week placebo-controlled trial of iloprost in patients with IPF and PH demonstrated no differences in 6 MW, NYHA functional class, Borg dyspnea score, or oxygen desaturation with exercise. 51
The value of inhaled prostanoid therapy has been demonstrated in the INCREASE trial comparing inhaled treprostinil with placebo and demonstrating a 31-m improvement in placebo-corrected peak 6 MW distance at 16 weeks. 52 Additional benefits included improvement in time to clinical worsening and lowering of N-terminal pro-B-type natriuretic protein (NT-proBNP) level. An unexpected finding in post hoc analysis was an improvement in forced vital capacity (FVC) at 16 weeks, especially in the subgroup with IPF where placebo-corrected FVC increased by 168 ml. 53 Post hoc analyses of the INCREASE trial revealed that patients across all ILD phenotypes experienced fewer multiple disease progression events with inhaled treprostinil compared to placebo. 54 Patients who achieved higher dosing levels of inhaled treprostinil experienced a lower incidence of clinical worsening and greater clinical improvement. 55 Data from the long-term extension study revealed stabilization of exercise tolerance and sustained improvement in FVC. 56
Inhaled treprostinil solution was initially approved for use in PAH in 2009, and based on the findings from the INCREASE trial, inhaled treprostinil solution was approved by the Federal Drug Administration (FDA) in 2021 to improve exercise capacity in patients with WHO Group 3 PH associated with ILD. Inhaled treprostinil solution is delivered with a portable, battery-operated, ultrasonic nebulizer (Optineb) from which patients typically inhale 9–12 breaths four times daily. Each breath provides approximately 6 mcg of treprostinil. 57 The nebulizer remains available, but since 2022 inhaled treprostinil has been available in a dry powder formulation that delivers a specified dosage of treprostinil with 1 inhalation four times daily. The specified dosage is provided by inserting a cassette with 16, 32, 48, or 64 mcg dry powder treprostinil into a small dry powder inhaler. The safety and effectiveness of the dry powder inhaler were demonstrated in the BREEZE study, a study comparing the dry powder inhaler with nebulized treprostinil. 58 In BREEZE, a group of patients stable on nebulized treprostinil transitioned to the dry powder inhaler at an equivalent dose level. Significant improvements in 6 MW distance and the PAH Symptoms and Impact questionnaire (PAH-SYMPACT) were noted after 3 weeks on the dry powder inhaler. Responses on the preference questionnaire for inhaled treprostinil devices indicated a preference for the dry powder device. The most common side effects with either inhaled treprostinil solution or dry powder formulation include cough, throat irritation or pain, headache, nausea, and flushing.52,58
Investigational inhaled therapies
Other inhaled treatment options are currently under investigation for the treatment of PH-ILD and may prove useful in the future. Inhaled treprostinil has been introduced in additional forms.59 –61 One alternative form of inhaled treprostinil, LIQ861, has been created with novel PRINT technology to create inhalation particles of uniform size, trefoil shape, and composition to optimize drug delivery. 59 This new formulation has been studied in PAH patients who were either prostacyclin-naïve or transitioned from nebulized treprostinil (INSPIRE Trial). The INSPIRE Trial was designed to evaluate the safety and tolerability of this alternative dry powder formulation with a primary emphasis on adverse events (AEs) and serious AEs. Adverse events included cough, throat irritation, and other side effects common to prostacyclin class agents; most were mild to moderate in severity. Analysis of secondary efficacy measures, such as 6MW, functional class, and NT pro-BNP levels, revealed stability or improvement after 1 year on treatment. The dosing and tolerability profile of LIQ861 is also under investigation in PH-ILD. An inhaled liposomal formulation of treprostinil (L606) has demonstrated extended plasma levels and sustained pharmacological effects in preclinical study 60 and is currently under phase III clinical investigation in patients with PAH and PH-ILD (NCT04691154). Treprostinil palmitil is yet another variation on inhaled treprostinil. 61 Treprostinil palmitil is a hexadecyl ester prodrug of treprostinil designed to provide prolonged delivery in the lung and thus has the potential for once daily dosing. 61 Clinical investigation of inhaled treprostinil palmitil in dry powder formulation is ongoing in patients with PAH (NCT05147805) and PH-ILD (NCT05649722).
Inhaled NO was approved by the FDA in 1999 for use in newborns with hypoxemic respiratory failure associated with pulmonary hypertension. 62 The development of portable NO generators has allowed the exploration of inhaled NO as a viable treatment option in PH-ILD. An exploratory study in a cohort of patients with pulmonary fibrosis at risk for pulmonary hypertension compared placebo with inhaled NO at 30 mg/kg/h over 8 weeks and demonstrated significant improvement in moderate-vigorous activity as measured by actigraphy. 63 This was followed by a longer term, phase II, placebo-controlled study in a second cohort of patients with inhaled NO delivered at a dose of 45 mg/kg/h over 4 months. 64 There was a notable placebo-corrected increase in moderate-vigorous activity tolerance in the treatment arm along with trends toward improvement in dyspnea and quality of life scores. 62 However, results from a phase III comparison of inhaled NO with placebo (REBUILD Trial) in patients with pulmonary fibrosis at risk for pulmonary hypertension did not meet the primary endpoint of change in moderate-to-vigorous physical activity as measured by actigraphy after 16 weeks of treatment leaving the future of inhaled NO for PH-ILD in question (NCT03267108).
Inhaled mosliciguat is an inhaled soluble guanylate cyclase (sGC) stimulator that targets heme-free apo-soluble guanylate cyclase. Intracellular heme-free apo-sGC develops as a consequence of oxidative stress in cardiopulmonary disease and is less responsive to nitric oxide and PDE-5 inhibitors. 65 Currently available sGC stimulators require heme-containing sGC to be effective. Inhaled mosliciguat has shown beneficial effects including reduction in pulmonary artery pressure and bronchodilation in animal models. 66 Another inhaled sGC stimulator, MK-5475, was associated with reduction in PVR in a phase I study 67 and is currently under investigation in patients with PH and COPD. The potential for inhaled sGC stimulators in treating PH-ILD is certainly intriguing, although will require future investigation in this disease state.
Platelet-derived growth factor (PDGF) signaling has been implicated in progenitor cell-dependent lung vascular neomuscularization and remodeling. 68 Tyrosine kinase inhibitors such as seralutinib inhibit PDGF and may be useful adjuncts to currently approved treatments for PAH and/or PH-ILD. Seralutinib is a potent inhibitor of platelet-derived growth factor receptor ∝/β (PDGFR∝/β), colony-stimulating factor I receptor and stem cell factor receptor (c-KIT) pathways involved in vascular remodeling. 69 A phase II placebo-controlled study of inhaled seralutinib (TORREY Trial) in PAH has concluded and achieved its primary endpoint with a significant reduction in PVR in patients receiving inhaled seralutinib. 70 A phase III study of inhaled seralutinib in PAH is underway (NCT05934526). Inhaled vardenafil is being evaluated for its use to relieve acute symptoms during exercise that are not being controlled with the patient’s long-term PAH treatment regimen. 71 If these novel inhaled therapies prove effective in PAH, hopefully, their potential for the treatment of PH-ILD will also be considered in future studies. A summary of currently available therapeutic options for PH-ILD with proven efficacy or under investigation is provided in Table 2.
Summary of currently available therapeutic options for PH-ILD with proven efficacy or under investigation.
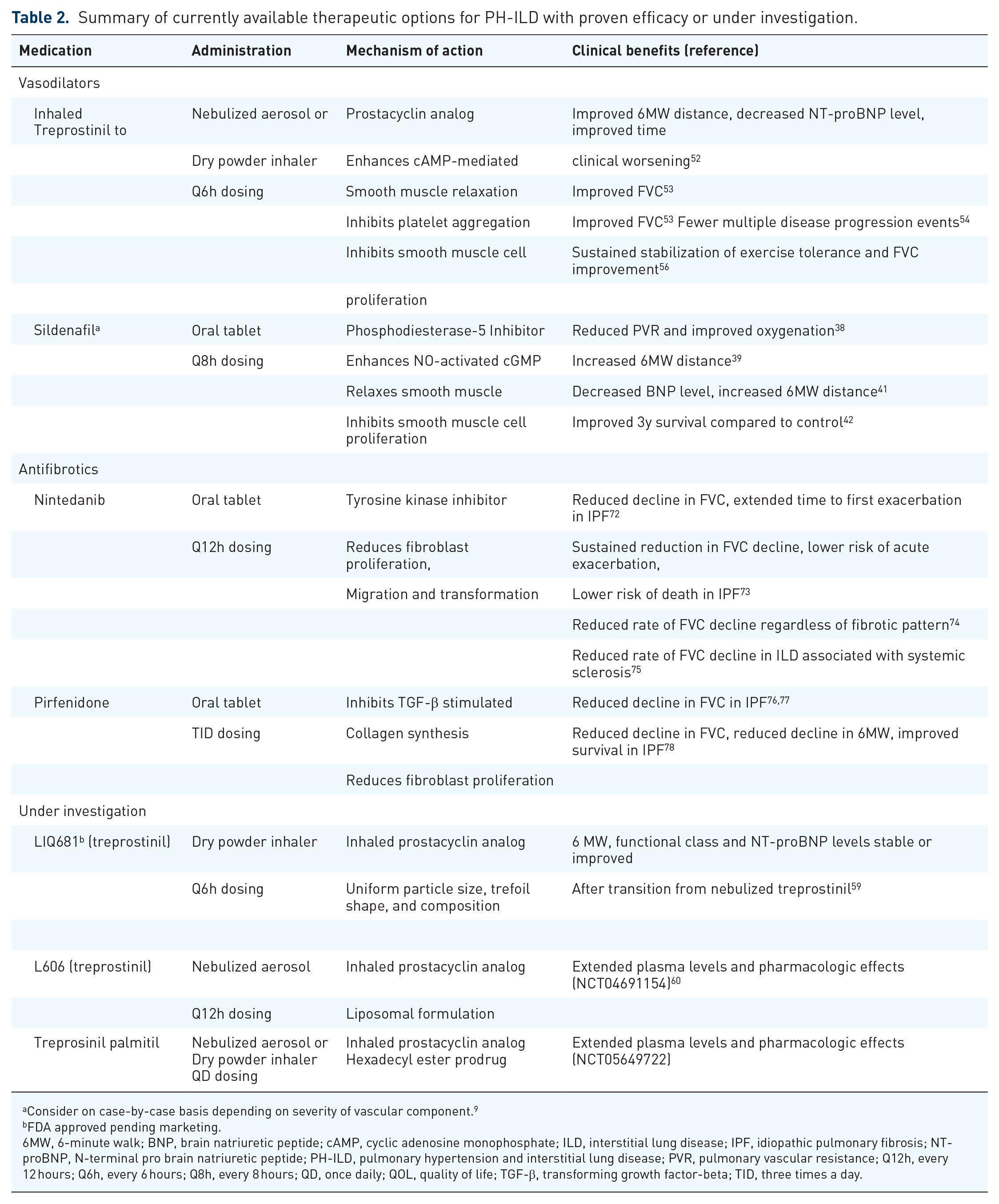
Consider on case-by-case basis depending on severity of vascular component. 9
FDA approved pending marketing.
6MW, 6-minute walk; BNP, brain natriuretic peptide; cAMP, cyclic adenosine monophosphate; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; NT-proBNP, N-terminal pro brain natriuretic peptide; PH-ILD, pulmonary hypertension and interstitial lung disease; PVR, pulmonary vascular resistance; Q12h, every 12 hours; Q6h, every 6 hours; Q8h, every 8 hours; QD, once daily; QOL, quality of life; TGF-β, transforming growth factor-beta; TID, three times a day.
Combination vasodilator–antifibrotic therapy in PH-ILD
To date, inhaled treprostinil is the only targeted pulmonary vasodilator that has shown promise for the treatment of pulmonary hypertension in patients with ILD. It would seem intuitive that combination therapy with a targeted pulmonary vasodilator and an antifibrotic, such as pirfenidone or nintedanib, could provide greater benefit than either approach alone in this disease affecting vascular and parenchymal compartments. Little has been reported about the effects of a combination of PAH and antifibrotic therapies in the ILD population. One investigation, the INSTAGE study, explored the value of adding sildenafil to nintedanib in patients with IPF and a DLCO < 35% predicted. 79 The study failed to achieve the primary endpoint of change in St George’s Respiratory Questionnaire at 12 weeks. However, there was a favorable improvement in FVC compared to placebo at 12 weeks. A longer term randomized, controlled trial of sildenafil added to pirfenidone failed to demonstrate improvement in time to clinical worsening. 80 Investigation of combination therapy options employing other pulmonary vasodilators is an area of unmet need in PH-ILD.
Approach to diagnosis of PH-ILD
We have previously highlighted the exceedingly poor survival prognosis in those with even mild PH and coexisting ILD, thereby underscoring the importance of early detection and treatment. Kimura et al. demonstrated not only a roughly 50% decrease in survival in patients with IPF and PH compared to IPF alone but also demonstrated similar rates of decline whether mPAP was 21–25 mmHg or was greater than 25 mmHg. 7 The vast majority of patients with PH-ILD have mild pulmonary hypertension with mPAP below the threshold at which distinct physical signs of PH are usually observed.6,81
Early detection of pulmonary hypertension in patients with ILD depends on awareness of clinical features indicating the coexistence of vascular disease (Table 3). Symptoms of pulmonary hypertension overlap with those of ILD and are of limited value in distinguishing the ILD patient with pulmonary hypertension. Pulmonary hypertension should be considered when symptoms worsen without corresponding changes in lung function or seem out of proportion to the severity of interstitial disease. Physical findings indicating PH may not be apparent in the ILD patient until vascular resistance is severe and associated with right heart failure. 3 Clinical markers that might indicate the presence of pulmonary hypertension include rising oxygen requirements or worsening desaturation with exercise in the absence of measurable changes in lung function, declining 6 MW distance with stable lung function, enlarged pulmonary artery or right ventricle on computed tomography (CT) imaging, elevated BNP or NT-proBNP levels, diffusion capacity for carbon monoxide (DLCO) <40%, or an increasing FVC/DLCO ratio.4,9,82 Parikh and colleagues reported an early detection tool with 86.5% sensitivity and 86.3% specificity for PH in ILD based on a score derived from 6-minute walk distance, physical exam findings, DLCO, supplemental oxygen use, BNP or NT-proBNP, syncope or presyncope, enlarged PA on CT, and presence of connective tissue disease or sarcoidosis. 83 Based on a retrospective analysis of data from IPF patients at initial evaluation, Furukawa and colleagues developed a simpler scoring system to predict the presence of PH based on DLCO <50% predicted, ratio of PA to aorta diameter on CT > 0.9 and arterial oxygen tension <80 Torr. 84

BNP, brain natriuretic peptide; CT, computed tomography; DLCO, diffusing capacity for carbon monoxide; ILD, interstitial lung disease; NT- pro BNP, N-terminal prohormone of brain natriuretic peptide.
Echocardiography is the most commonly used screening tool for PH, but accuracy is often limited in patients with lung disease. The triscupid regurgitant jet velocity (TRV) used to estimate right ventricular systolic pressure (RVSP) can be difficult to visualize and prevents determination of the tricuspid regurgitant velocity in 50% or more of ILD patients suspected of having PH. 9 Further, echocardiography has been shown to under- or overestimate the RVSP by more than 10 mmHg compared to right heart catheterization in about half of patients with ILD. 86 In a study of ILD patients suspected of having PH, 40% of those with a low probability of having PH by echocardiogram had PH confirmed by right heart catheterization. 87 Other echocardiographic measures may be more accurate than TRV in this population including right atrial area, RV outflow tract diameter, RV:LV ratio, LV eccentricity index, and tricuspid annular plane excursion.88 –90 Machine learning algorithms have been shown useful in integrating several echocardiographic parameters to predict pulmonary hypertension and may enhance the ability to more accurately detect early stages of PH-ILD.85,91
Detailed structural and hemodynamic information for screening and diagnosis of PH-ILD may be attainable with cardiac MRI (cMRI). Magnetic resonance imaging can assess morphological changes in the heart, pulmonary artery diameter and wall thickness, measures of RV systolic and end-diastolic volumes, and septal bowing. 92 4D MRI can provide qualitative and quantitative assessments of vascular hemodynamics which may serve as a basis for RHC in patients with borderline measures on transthoracic echocardiography. 93 In a prospective cohort of patients with chronic lung disease, Alkhanfar and colleagues diagnosed PH by MRI with 92.3% sensitivity and 70.2% specificity. 94 Although not widely available, cMRI is certainly a promising tool for early diagnosis of PH-ILD.
In those patients suspected of having pulmonary hypertension, whether on the basis of echocardiography or other clinical markers, right heart catheterization is required for definitive diagnosis and assessment of severity. Prior to the 2022 ESC/ERS guidelines for diagnosis of PH, diagnosis by RHC relied on a mPAP > 25 mmHg and PVR > 3WU. Based on that definition, PH in patients with lung disease has generally been considered significant if mean PAP > 35 mmHg or mean PAP > 25 mmHg with PVR > 4WU. 95 Many ILD patients with mild PH may not meet these older hemodynamic criteria yet are known to have unfavorable outcomes similar to those with more severe disease. 10 Benefits of treating PH in ILD determined by newer hemodynamic parameters remain to be determined.
Adjunctive measures
General measures
Even considering the benefits with newer antifibrotic therapies for fibrotic lung disease and inhaled treprostinil for the treatment of PH in patients with PH-ILD, several adjunctive measures are important components of a comprehensive treatment plan (Table 4). It is important to optimize the treatment of comorbidities that may affect symptoms or management of PH-ILD. Diuretics may be indicated to control fluid overload in those patients with significant right ventricular dysfunction. Nutritional referral may be helpful for guidance on limiting sodium intake and maintenance of optimal calorie balance.
Adjunctive measures for patients with PH-ILD.

PH-ILD, pulmonary hypertension and interstitial lung disease.
Immunizations
Preventive immunizations against pneumococcal pneumonia, influenza, and SARS-CoV-2 are recommended. 9 Patients with PH-ILD should have annual immunization against influenza virus, assuming they have no allergy to vaccine components. There are several vaccines against pneumococcal pneumonia available, each covering different serotypes of Streptococcus pneumoniae. 96 There are two basic types of pneumococcal vaccine; pneumococcal conjugate (PCV) and pneumococcal polysaccharide (PPSV). Patients receiving the PCV15 vaccine should be vaccinated with PPSV23 1 year later. Patients receiving the PCV 20 or PCV21 vaccine need no additional pneumococcal vaccine. Patients with chronic respiratory disease are also at higher risk for respiratory syncytial virus (RSV) infection, and the Center for Disease Control (CDC) recommends RSV vaccination in adults aged 60–74 with increased risk of severe infection. 97
Smoking cessation
Smoking is known to induce PAH in animal models. For example, mice exposed to inhaled nicotine demonstrate the development of pulmonary hypertension accompanied by vascular and right heart remodeling. 98 Active cigarette smoking or exposure to second-hand smoke was associated with higher prevalence of PAH in two case–control studies, although a causative relationship has not been proven in humans.99,100 Endothelin and its receptors have been linked to the development of pulmonary hypertension in chronic lung disease associated with cigarette smoking. 101 Smoking has been associated with an increased risk of developing IPF in humans.102,103 In a cohort study of patients with IPF, lifelong nonsmokers experienced better survival outcomes compared to those who were former or current smokers. 104 Smoking cessation should be recommended, and smoking cessation aids or counseling should be provided for patients to promote success. 105
Oxygen therapy
Hypoxemia not only portends risk of organ dysfunction further complicating vascular disease and ILD but it also causes breathlessness, impaired activity tolerance, and reduced quality of life. Oxygen requirements should be evaluated on a regular basis and supplemental oxygen provided with the aim of limiting symptoms and vasoconstriction. Oxygen needs should be assessed at rest, with activity and while sleeping. Oxygen therapy has been recommended for those with COPD and ILD who have severe hypoxemia defined as PaO2 < 55 mmHg or SpO2 < 88% by pulse oximetry, or alternatively if PaO2 = 56–59 mmHg or SpO2 = 89% with edema, hematocrit >55% or P pulmonale on electrocardiogram. 106 American Thoracic Society (ATS) guidelines recommend long-term oxygen therapy for at least 15 h/day when patients have resting room air hypoxemia and for patients with exertional room air hypoxemia. 106 The guidelines also recommend considering liquid portable oxygen for patients who are active outside the home and require more than 3 L/min with exertion. There are a variety of portable concentrators available that improve mobility in patients with low flow requirements. Patients with nocturnal hypoxemia should be screened for sleep-disordered breathing, as supportive ventilation may be required to correct hypoxemia. 107
Pulmonary rehabilitation
Structured pulmonary rehabilitation has proven beneficial in PAH and in PH-ILD with noted improvements in oxygen consumption, functional capacity, and quality of life.108,109 Pulmonary rehabilitation (PR) improves the quality of life in chronic lung disease by providing disease education, nutritional counseling, individualized endurance, and resistance exercise prescription and monitoring while determining supplemental oxygen needs with different levels of activity. Unfortunately, availability is limited for many patients, due to a paucity of center-based programs, travel distance, and health insurance reimbursement. 110 A variety of commercially available virtual PR programs are available for patients with limited access to centers where PR is offered. Telehealth PR is included as an option in recently updated ATS pulmonary rehabilitation guidelines based on evidence supporting improvements in 6 MW, symptoms, and quality of life similar to traditional center-based programs. 111 Caution is advised in recommending telehealth pulmonary rehabilitation due to variability in patient assessment, prescription standards, monitoring, and supervision. 112 Furthermore, there are questions about the safety of telehealth programs for patients with comorbidities, such as heart failure, or who have high oxygen requirements. 113 Although virtual pulmonary rehabilitation may offer similar benefits to center-based PR for patients with stable chronic lung disease, in particular COPD, the effectiveness of telehealth PR in pulmonary hypertension and ILD remains to be established.
Collaborative care
International guidelines have recommended a multidisciplinary approach to the diagnosis of IPF including pulmonologists, radiologists, pathologists, and in some cases rheumatologists. 114 The high level of diagnostic confidence achievable with this approach has gained popularity for the evaluation of interstitial lung diseases in general. 115 Since PH-ILD encompasses vascular and parenchymal disease, it makes sense to employ this collaborative approach, not only for the purpose of diagnosis but also for treatment planning and supportive measures. While accurate diagnosis may depend primarily on input from pulmonologists, radiologists, and pathologists, treatment and supportive management beyond diagnosis may be best accomplished through collaboration between clinicians, nutritionists, psychologists, social workers, and PR experts. Including a palliative care team can provide psychosocial support and assist with symptom control throughout the course of PH-ILD treatment. Likewise, patients can experience psychosocial benefits from shared experiences with peers in patient support group settings. When local resources and expertise are limited, early collaboration with an expert center can provide useful input in accurate diagnosis, unique treatment options, clinical trial opportunities, and expedited referral for lung transplantation.116,117
Lung transplantation
Lung transplantation should be considered early for patients with PH-ILD. Inadequate response to initial therapy, rapid disease progression, and confirmed or suspected pulmonary veno-occlusive disease are triggers for lung transplant referral in patients with PH. 118 Referral for lung transplantation is recommended when any patient with ILD has pulmonary hypertension. 119 Specific indications for referral in patients with ILD include the presence of usual interstitial pneumonia, connective tissue disease or familial pulmonary fibrosis, and progression of inflammatory ILD despite treatment. Deteriorating lung function (absolute fall in FVC > 10%, DLCO > 15%) or activity tolerance (decrease in 6MW 50 m or more), and hospitalization for exacerbation should also prompt referral to a lung transplant program. 119
Pulmonary hypertension increases the risk of complications and mortality when patients undergo lung transplant for ILD. 120 Bilateral lung transplantation is performed for primary PH and for ILD when PH is more severe (mPAP > 40 mmHg), although single lung transplant may be a viable option in PH-ILD with less severe pulmonary hypertension.121,122
Patient-reported outcome measures
While clinical trial data provide quantitative indicators of significant change in clinical variables associated with a disease state, patient-reported outcome measures (PROMs) may provide a different perspective on the real-world impact of a particular treatment.123,124 At the individual patient level, PROMs have value in monitoring treatment effects and guiding modifications in the therapeutic approach. In clinical trials, PROMs provide useful insight into the impact of investigational agents on patient quality of life. PROMs are not only useful on an individual patient level but they may also provide valuable effectiveness data at organizational levels where policy is generated. PROMs may be very useful in guiding research and patient care decisions in chronic, progressive diseases, such as PH-ILD, where there is a great need for the development of effective treatment options and where preservation of quality of life may drive a patient’s tolerance for available treatment options.
Conclusion
WHO Group 3 pulmonary hypertension carries the least favorable survival prognosis when compared to other WHO Groups, with the greatest survival disadvantage in those with PH-ILD. Although pulmonary hypertension in patients with fibrotic lung disorders has traditionally been thought to arise from the effects of parenchymal destruction and resulting hypoxemic vasoconstriction, there is evidence that mediators arising from endothelial dysfunction and disordered intracellular signaling pathways are involved. Unlike WHO Group 1 PAH, far fewer options have been discovered for the treatment of PH-ILD. Inhaled treprostinil has been shown to positively impact functional capacity and delay clinical worsening in patients with PH-ILD, and newer antifibrotic therapies have demonstrated the ability to slow the progression of parenchymal injury. More information is needed about the benefits of combining pulmonary antihypertensive and antifibrotic therapies in these patients. Given the exceedingly poor prognosis in PH-ILD, the importance of early diagnosis and treatment cannot be over-emphasized. More effective management of PH-ILD can be achieved with a collaborative approach including not only essential clinicians but also experts in nutrition, PR, social services, palliative care, and expert centers that can offer clinical trial opportunities and lung transplantation. New inhaled therapies are under investigation for PAH and need to be studied more intently in PH-ILD. Much more investigation in search of cellular and molecular mechanisms of disease is needed in order to identify pathways that will lead to more effective treatment options for this disease state.