
Abstract
Interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH) are leading causes of morbidity and mortality in systemic sclerosis (SSc). As symptoms are often under-reported in SSc, early screening of ILD and PAH is of paramount importance, and early treatment may be associated with better clinical outcomes. Serologies are particularly helpful in identifying patients at risk for pulmonary involvement. Pulmonary function testing, high-resolution computed tomography of the chest and echocardiography are important tools in the initial screening of these patients. Extensive research has also led to an improved understanding of the mediators involved in the pathogenesis of ILD and PAH. As a result, there have been significant advances in the development of novel targeted therapeutics and an increase in the number of early-phase clinical trials in SSc.
Systemic sclerosis (SSc) is a heterogeneous autoimmune disease characterized by vasculopathy and progressive fibrosis with multiorgan involvement. Interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH) are the most common forms of lung involvement and are the leading causes of mortality in patients with SSc. 1 Effective therapies for both SSc-ILD and SSc-PAH have historically been limited. Fortunately, in recent years, there has been significant progress in understanding the pathogenesis and in the development of novel therapies for ILD and PAH.
Interstitial lung disease
Background and epidemiology
Interstitial lung disease is the leading cause of mortality in SSc. 2 Clinically relevant pulmonary fibrosis is present in approximately 25% of all SSc patients, 3 but up to 90% of patients with SSc show changes consistent with ILD on high-resolution computed tomography (HRCT) of the chest, and between 40–75% of patients exhibit some pulmonary function test (PFT) abnormality.4–6
Patients with SSc are classified as limited cutaneous SSc (lcSSc) or diffuse cutaneous SSc (dcSSc) based on the extent of skin involvement. Patients with lcSSc often develop skin involvement of the face and skin distal to the elbows and knees, and dcSSc patients have more extensive skin involvement including the trunk and extremities. Patients with dcSSc or Scl-70 (anti-topoisomerase I) antibodies are more likely to develop ILD, whereas patients with lcSSc or anticentromere antibodies tend to have a lower prevalence of pulmonary fibrosis. It has been suggested that the auto-antibody profile is of particular importance in SSc-ILD, more so than just the clinical subset of the disease.
The antinuclear antibody (ANA) is present in approximately 90–95% of patients with SSc. 7 Specific ANA patterns can be useful in predicting clinical phenotypes. In a large European scleroderma database, 60% of patients with positive Scl-70 (anti-topoisomerase I) antibodies had ILD, compared with 21% of patients with anticentromere antibodies. 8 Other factors, such as modified Rodnan Skin Score (mRSS), African-American ethnicity, creatine phosphokinase levels, creatinine levels and cardiac involvement are also independently associated with ILD in patients with SSc. 3
Diagnosis and screening
The multiorgan system involvement of SSc makes it particularly difficult to distinguish specific symptoms in patients. Patients with mild ILD may be asymptomatic at early stages of the disease, and often develop fatigue, dyspnea on exertion and cough as the extent of pulmonary fibrosis progresses. Given the importance of identifying and treating ILD, early screening and diagnosis is of paramount importance even when minimally symptomatic. PFTs and HRCT of the chest play important roles in both the diagnosis and determination of disease severity. The forced vital capacity (FVC), diffusing capacity for carbon monoxide (DLCO), and total lung capacity (TLC) are important parameters to consider in the evaluation of SSc-ILD, and the FVC, in combination with the extent of disease on HRCT can help stratify patients for treatment. 9 In a patient without respiratory symptoms, FVC and DLCO > 80% predicted are usually considered normal; however, in a patient with respiratory symptoms, an FVC or DLCO > 80% could represent a significant decline from a previous value and should be further evaluated. SSc-ILD is typically characterized by a restrictive ventilatory defect with FVC < 70% predicted and an forced expiratory volume in 1 second (FEV1):FVC ratio of >0.8. Both FVC and DLCO are prognostic factors in patients with SSc-ILD. Among 80 patients who underwent lung biopsy for SSc with fibrosing alveolitis, lower initial DLCO and FVC were associated with mortality. 10
HRCT is often used in conjunction with PFTs to determine the pattern and extent of involvement. The most common pattern seen on HRCT is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can also be seen in up to 40% of cases.10,11 The NSIP pattern on HRCT manifests as ground glass opacities (GGOs) in a peripheral distribution with subpleural and basilar predominance (Figure 1A). In more severe disease, volume loss with a reticular pattern and traction bronchiectasis can also be seen. In UIP, HRCT findings include reticulonodular opacities, traction bronchiectasis and honeycomb cysts (Figure 1B). A normal chest computed topography (CT) at baseline is generally reassuring; in one study, 85% of SSc patients who had a normal HRCT at baseline still had a normal HRCT at 5 years. 12
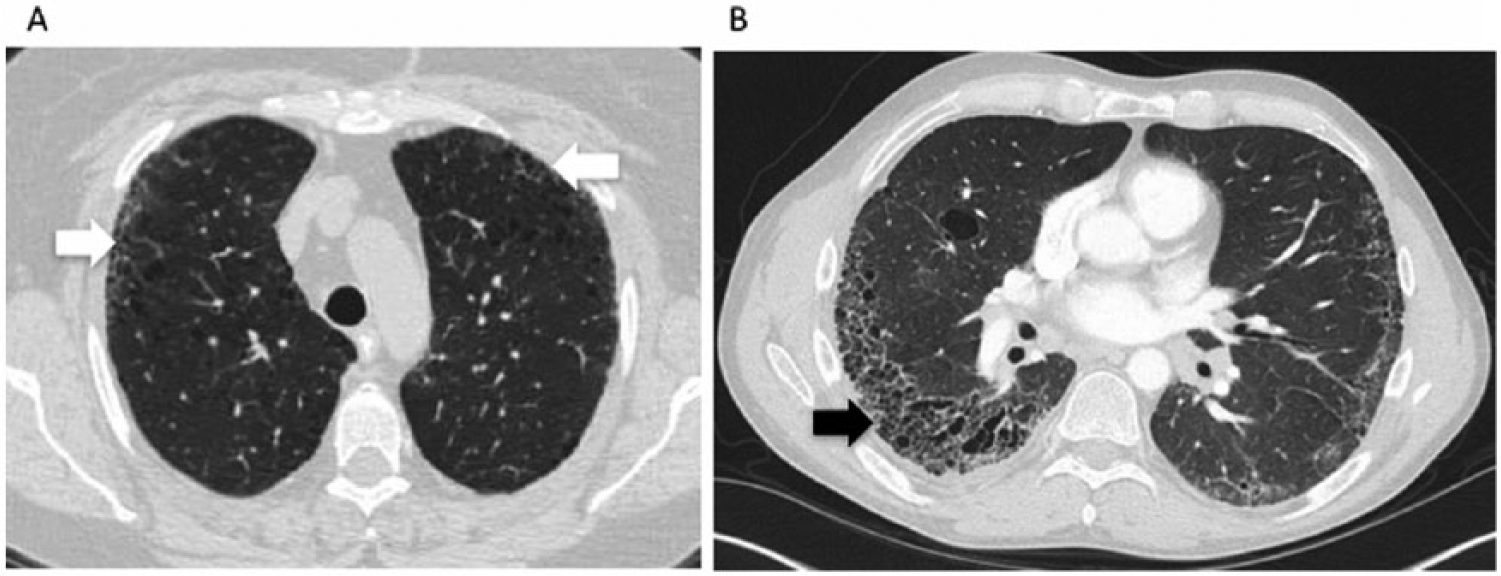
Characteristic radiographic findings on high-resolution computed tomography. (A) Subpleural ground glass opacities (white arrows) and traction bronchiectasis seen in nonspecific interstitial pneumonia, and (B) honeycombing (black arrow), and bronchiectasis seen in usual interstitial pneumonia.
Both HRCT and PFTs are well-accepted methods for the diagnosis of SSc-ILD, with 100% and 99% consensus among SSc experts for the use of HRCT and PFTs, respectively. 13 As HRCT may capture minor interstitial changes of uncertain significance, and PFTs may be confounded by the presence of other pulmonary abnormalities such as emphysema, pulmonary hypertension (PH), muscle weakness or pleural disease, the combination of HRCT, PFTs and respiratory symptoms are superior for accurately diagnosing SSc-ILD. Annual PFTs are recommended to screen patients for SSc-ILD. Abnormalities on PFTs should prompt an HRCT. Some have suggested that HRCT be performed in all patients with SSc as a screening exam and if abnormal be followed by PFTs every 3–6 months depending on symptom progression. 14
The 6-min walk test (6MWT), a measure used to assess a patient’s submaximal exercise capacity over a flat surface over 6 min, has been an important tool in evaluating idiopathic pulmonary fibrosis (IPF). In SSc, however, the value is less reliable, as patients may have limitations due to musculoskeletal involvement. Nonetheless, the presence of a major oxygen desaturation with ambulation should not be overlooked, as it may indicate the presence of either ILD or PH.
Current therapies
Given the potential toxicities of many of the medications used to treat SSc-ILD, the decision of which patients to treat must be considered carefully. One algorithm based on FVC and the extent of involvement on HRCT has been proposed to help determine which patients may benefit from treatment. In this algorithm, HRCT involvement of >20% and FVC < 70% were associated with increased mortality risk, and these parameters, in addition to short disease duration and evidence of recent progression may help guide clinicians in deciding which patients to treat. 9
Immunosuppressive therapy remains the best studied and most commonly used treatment for SSc-ILD (Table 1). Cyclophosphamide and mycophenolate mofetil (MMF) have both been studied in randomized controlled trials. In the Scleroderma Lung Study I (SLS I), a multicenter, double-blind randomized controlled trial (RCT), treatment with 1 year of oral cyclophosphamide was compared with placebo, and change in FVC was measured as the primary outcome. 11 At 1 year, patients treated with cyclophosphamide experienced a modest improvement in FVC compared with those treated with placebo. At 2 years, however, this effect was gone. Patients treated with cyclophosphamide also had improved dyspnea, total lung capacity and modified Rodnan Skin Score (mRSS), a validated score used to assess skin thickening in patients across 17 different areas. In another RCT, 6 months of corticosteroids and intravenous (IV) cyclophosphamide followed by azathioprine for 6 months was compared with placebo in patients with SSc-ILD. 15 Patients in the treatment group showed a trend towards improved FVC compared with the placebo group, although this did not reach statistical significance, perhaps due to the small sample size. Based on indirect comparisons of IV versus oral cyclophosphamide in this and other studies, IV cyclophosphamide may be associated with fewer adverse effects (i.e. risk of bladder cancer), with similar efficacy. However, given the potential serious adverse effects of cyclophosphamide including infection, hemorrhagic cystitis and secondary malignancy, alternative treatment options remain appealing.
Current treatment options for systemic sclerosis with interstitial lung disease.

DNA, deoxyribonucleic acid; FVC, forced vital capacity; RCT, randomized controlled trial; DLCO, diffusing capacity for carbon monoxide; N/A, not applicable; SSc-ILD, systemic sclerosis with interstitial lung disease.
MMF has emerged as an alternative agent to treat SSc-ILD with a more favorable safety profile. The Scleroderma Lung Study II, a multicenter, double-blind RCT compared 2 years of MMF with 1 year of oral cyclophosphamide followed by 1 year of placebo for the treatment of SSc-ILD. 16 A total of 142 patients were randomized, and the primary endpoint was a change in FVC as a percentage of the predicted normal value (FVC %) over 2 years. Changes in FVC at 2 years in the MMF group were non-inferior to those in the cyclophosphamide group. Significantly fewer patients discontinued their medication in the MMF group, and adverse events such as weight loss and leukopenia/thrombocytopenia were less common in the MMF group. Overall, these results provide evidence that MMF is a reasonable first-line therapy for SSc-ILD, with potentially fewer side effects than cyclophosphamide, though both MMF and cyclophosphamide are effective for SSc-ILD.
The evidence for use of azathioprine in SSc-ILD is less robust. In a retrospective study of 11 patients with SSc-ILD, treatment with azathioprine resulted in stable FVC and dyspnea index scores in 8 patients. 19 In another study of SSc-ILD, treatment with 6 months of monthly IV cyclophosphamide followed by 18 months of azathioprine resulted in stable or improved PFTs at 6 months in 70% of patients, but the specific role of azathioprine in this outcome is unclear. 20 Other studies have suggested that azathioprine may actually be harmful; a randomized trial of cyclophosphamide versus azathioprine in the treatment of early diffuse SSc demonstrated that FVC and DLCO worsened in the azathioprine group. 21
Although glucocorticoids have been used in combination with other immunosuppressive agents such as cyclophosphamide and azathioprine in smaller trials of SSc-ILD, the role for high-dose steroids in SSc-ILD is generally limited. In addition, given the increased risk of scleroderma renal crisis associated with prednisone at doses of >15 mg/day, there is concern with using prolonged high doses of prednisone in patients with SSc.22,23
Historically, lung transplantation in SSc has been difficult, given the frequent gastrointestinal (GI) involvement in these patients. Both reflux and esophageal dysmotility raise concern for recurrent aspiration events and increased lung damage in the allograft. However, studies comparing outcomes after lung transplant in SSc-ILD patients with other ILD patients have shown similar 1- and 5-year survival rates.17,18 Lung transplant remains an important consideration for patients with severe or rapidly progressive disease that does not respond to other treatment modalities.
Experimental therapies
Traditional immunosuppressive therapies remain the mainstay of treatment, with the primary goal of disease stabilization. Although many of the current treatments result in stabilization or a modest improvement in FVC, these effects may not be sustained, as demonstrated in SLS I. Hence, there is great interest in the development of more targeted therapies for SSc-ILD that may result in more significant and sustained improvements in lung function (Table 2). As the molecular pathways involved in SSc and fibrosis are further elucidated, the opportunity for development of targeted therapies has increased.
Investigational treatments for systemic sclerosis with interstitial lung disease.

FVC, forced vital capacity; RCT, randomized controlled trial; IPF, idiopathic pulmonary fibrosis; TGF-β, transforming growth factor-beta; mRSS, modified Rodnan Skin Score; IL-6, interleukin-6; LPA, lysophosphatidic acid; SSc-ILD, systemic sclerosis with interstitial lung disease; CD20, a B-lymphocyte antigen.
Fresolimumab/transforming growth factor-beta pathway
One of the key mediators in SSc-ILD, and in all fibrotic processes, is the pleotropic cytokine, transforming growth factor-beta (TGF-β). Given the important role of TGF-β in fibrosis, it is a natural target for therapy in SSc. Fresolimumab, a monoclonal antibody to TGF-β that neutralizes all isoforms of the molecule, has been studied in a recent small open-label trial. 24 Two different doses of fresolimumab were compared in patients with early (<2 years) dcSSc with the primary outcomes being a change in mRSS, as well as a change in mRNA levels of two skin biomarkers, cartilage oligomeric protein (COMP) and thrombospondin-1 (THBS1) at 24 weeks. Both treatment groups showed a relative rapid decrease in mRSS and THBS1, suggesting that selective targeting of the TGF-β pathway may be beneficial in the treatment of patients with SSc. Larger studies are needed to determine the long-term effectiveness of this drug and whether it will be useful in SSc-ILD. Other drugs targeting the TGF-β pathway are also in development. Abituzumab, a monoclonal antibody against αv integrin, the integrin crucial for activation of latent TGF-β, will be examined in SSc-ILD in an upcoming phase II trial [ClinicalTrials.gov identifier: NCT02745145].
Other antifibrotic therapies
Two oral antifibrotic medications, pirfenidone and nintedanib, have recently been approved for use in IPF.28,35 Both medications slowed disease progression, as measured by FVC. An open-label phase II trial of pirfenidone in SSc-ILD (LOTUSS) was completed and demonstrated acceptable tolerability, even with over 60% of patients concurrently taking MMF, and a phase III trial is currently being planned. 27 A phase III double-blind, randomized controlled trial of nintedanib in SSc-ILD is currently underway and will also allow for the concurrent use of MMF [ClinicalTrials.gov identifier: NCT02597933]. The primary endpoint of this trial is a change in FVC annually over 52 weeks.
Tocilizumab
Tocilizumab, a monoclonal antibody against the IL-6 receptor, is currently approved for use in rheumatoid arthritis (RA) and is being evaluated in SSc. Based on preliminary data suggesting that IL-6 levels may be associated with DLCO and FVC decline, as well as death in SSc-ILD, 36 and that IL-6 levels are elevated in the serum of patients with SSc, 37 a phase II trial of tocilizumab in SSc was completed. 25 The primary endpoint was a reduction in mRSS at week 24. While this endpoint was not met, there was a trend towards improvement in skin scores in the tocilizumab group compared with the placebo group at week 48. In addition, there was a trend towards a slower decline in FVC in the tocilizumab group, although the study was not specifically designed to examine this endpoint. A phase III trial is currently being planned; whether IL-6 blockade will be useful for treatment of SSc-ILD remains to be determined.
Rituximab
Rituximab, a monoclonal antibody against CD20 (a B-lymphocyte antigen) used in the treatment of rheumatoid arthritis (RA)- and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, has come of interest in the treatment of SSc-ILD. In a small nested case-control study, SSc patients treated with rituximab versus matched controls (not treated with rituximab) showed improvement in mRSS, and in a small subset of patients with ILD there was no further decline in FVC. 30 Based on this study and other smaller studies, 29 there is hope that rituximab may be beneficial in SSc-ILD, but larger, randomized studies are needed.
Lysophosphatidic acid
Lysophosphatidic acid (LPA) is a small bioactive lipid mediator that has been shown to be important in the development of both pulmonary and dermal fibrosis in mouse models.38,39 Based on this preclinical work, a phase II trial of an LPA1 receptor antagonist was completed and demonstrated an excellent safety profile and promising clinical efficacy with regards to reduction in mRSS. 31 Patients with SSc-ILD were not specifically examined, and further studies are needed to determine whether targeting the LPA pathway may provide benefit in patients with SSc-ILD.
Autologous stem cell transplant
Given the relatively poor prognosis of SSc-ILD and the historic lack of targeted therapies for the disease, autologous hematopoietic stem cell transplant (HSCT) presents a potentially attractive therapeutic option. Three major trials have compared HSCT with cyclophosphamide in SSc patients with internal organ involvement. ASSIST (Autologous Stem Cell Systemic Sclerosis Immune Suppression Trial), a single-center study, found that patients with dcSSc and internal organ involvement treated with HSCT compared with IV cyclophosphamide for 6 months had improvement in skin scores and FVC at 12 months. 32 The ASTIS (Autologous Stem cell Transplantation International Scleroderma) trial, a multicenter study in Europe and Canada, found that dcSSc patients treated with HSCT had improved event-free and overall survival despite a 10% treatment-related mortality in the HSCT group, when compared with 1 year of IV cyclophosphamide. 33 The SCOT (Scleroderma: Cyclophosphamide Or Transplantation) trial, a multicenter study in North America, comparing IV cyclophosphamide for 1 year with HSCT, found that there was long-term superiority to myeloablative HSCT with lower transplant-related mortality than expected (3%). 34 HSCT may be considered for patients with severe disease who have been refractory to other treatment options.
Pulmonary hypertension
Background and epidemiology
PH is another leading cause of morbidity and mortality among patients with SSc. The World Health Organization (WHO) has classified patients with PH into five categories based on the etiology of PH (Table 3). Although patients with SSc are susceptible to all forms of PH, WHO Group 1, PAH, is the most common type of PH affecting patients with SSc. Other types of PH, including Group 2, PH due to left heart disease, and Group 3, PH due to lung disease are also common, given the prevalence of heart disease and ILD in patients with SSc. The presence of PAH is defined by a mean pulmonary arterial pressure (PAP) of >25 mmHg with a pulmonary capillary wedge pressure (PCWP) < 15 mmHg on right heart catheterization (RHC). The prevalence of PH in patients with SSc has ranged between 5 and 13% in various studies,40–42 and SSc patients with PAH are more likely to have a positive anticentromere antibody than those without PAH, though PAH is present in both diffuse and limited subsets of the disease. 43
World Health Organization clinical classification of pulmonary hypertension.

BMPR2, bone morphogenic protein receptor type II; ALK-1, activin-like kinase type I; ENG, endoglin; CAV1, caveolin-1; KCNK3, potassium-channel subfamily K-member 3; SMAD9, ;HIV, human immunodeficiency virus; CTD, connective tissue disease; PH, pulmonary hypertension.
The PHAROS (Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma) cohort has provided valuable information regarding clinical outcomes of patients with SSc-PAH. The PHAROS registry comprises two groups: (1) subjects with incident PH based on a RHC-confirmed diagnosis within 6 months of enrollment in the registry, and (2) subjects with increased risk of developing PAH, or ‘pre-PAH’. Subjects in the PH-confirmed group are then further classified by their WHO group designation. In a study of 131 patients from the PHAROS registry with RHC-proven PAH, 1-, 2- and 3-year survival rates were 93%, 88% and 75%, respectively. 44 A meta-analysis of 22 studies found 1-, 2-, and 3-year survival rates for SSc patients with PAH of 81%, 64% and 52% respectively. 45 The discrepancy between these studies may stem from the fact that patients in the PHAROS registry all had incident disease and therefore, on average, had lower NYHA (New York Heart Association) functional class (FC) than patients in other studies. Risk factors associated with a worse prognosis in SSc-PAH that were identified in the PHAROS study include age > 60, male sex, WHO functional class (FC) IV, and a DLCO < 39%. 44 The REVEAL (Registry to Evaluate Early and Long-Term PAH Disease Management) registry, a multicenter, prospective US-based registry of patients with a previous and new diagnosis (within 90 days) of PAH by RHC, reported that in addition to male sex and age > 60, other risk factors for mortality included systolic blood pressure ⩽ 110 mmHg, 6-min walk distance < 165 m, mean right atrial pressure > 20 mmHg, and pulmonary vascular resistance > 32 Wood units. 46
Diagnosis and screening
Given the lack of specific signs or symptoms associated with PAH, it can be difficult to determine which patients with SSc may have or be at risk for PAH. Although dyspnea, particularly on exertion, is a common symptom associated with PAH, nearly a quarter of patients with PAH did not exhibit dyspnea in one study, 43 and in another study, dyspnea did not differentiate between patients with or without PAH. 47 Given this lack of clear symptomatology, and the high morbidity and mortality associated with untreated PAH, accurate screening modalities are an important consideration.
Expert recommendations suggest that all patients with SSc be screened for PAH with PFTs that include single-breath DLCO, transthoracic echo (TTE) and N-terminal pro-brain natriuretic peptide (NT-pro-BNP). 48 Furthermore, PFTs and TTE should be performed on an annual basis, and more frequently in the setting of new signs or symptoms. Various professional societies have also offered recommendations for PAH screening for patients with SSc. The American College of Chest Physicians (ACCP) recommends that patients with SSc be monitored for the development of PAH, but does not specify the modality or frequency of screening. 49 The European Society of Cardiology (ESC)/European Respiratory Society (ERS) recommends TTE as a screening test in all patients with SSc, even those who are asymptomatic. 50 In addition, it suggests that annual screening with TTE, PFTs and biomarkers be considered, and SSc patients with a mean PAP between 21 and 24 mmHg should be closely monitored given their increased risk of PAH. The American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) recommends yearly TTEs and subsequent RHC if TTE demonstrates high right ventricular systolic pressure (RVSP) or right chamber enlargement. 51
Although TTE is often recommended as a screening tool for PH, echocardiography alone can often be inaccurate in estimating PAP, with both underestimation and overestimation of the actual value. 52 As a result, an effort to develop more accurate, yet still feasible, screening techniques is underway. In the DETECT study, 466 SSc patients (disease duration > 3 years) at increased risk for PAH underwent non-invasive testing followed by RHC to determine which variables should be included in an algorithm to screen SSc patients for PAH. 47 Based on multivariable analysis, a two-step screening process was developed, allowing TTE to be limited to those patients who are truly at increased risk for PAH. Six nonechocardiogram variables were used to determine a risk score, including FVC:DLCO ratio, presence of telangiectasias, positive anticentromere antibody, serum NT-pro-BNP level, serum uric acid level and the presence of right-axis deviation on electrocardiogram (EKG). If patients had a total risk score of >300, they were referred for TTE. Two additional echo variables, right-atrial area and tricuspid-regurgitant jet velocity were then used to determine referral for RHC. This algorithm resulted in 96% sensitivity and 98% negative predictive value with earlier detection of PAH. However, it is important to note that DETECT studied a high-risk SSc population (DLCO < 60%), and did not include patients with significant ILD. While it may be helpful in high-risk patients who are >3 years from disease onset, it may need to be modified in patients with ILD or concurrent emphysema, 53 and cannot be generalized to screen all patients with SSc. Additionally, DETECT does not provide guidance on the frequency of screening.
Current therapies
The WHO has developed a functional classification system to categorize PAH according to its clinical symptoms and severity (Table 4). 50 In general, oral therapy is recommended for patients with FC II or higher, and the goal of treatment is to improve FC. Current therapies for SSc-PAH target the vasoconstrictive and vasodilatory mediators produced by the dysfunctional endothelium. The three main targeted pathways for the treatment of SSc-PAH include the nitric oxide pathway, the endothelin pathway and the prostacyclin pathway. It is important to note that many of the large randomized trials in PAH have not been designed or powered to look at SSc-PAH specifically, and evidence for the use of these medications for SSc-PAH is gathered from subgroup analyses of larger studies or smaller series of SSc patients (Table 5).
World Health Organization functional class for pulmonary arterial hypertension.

Medications and trials for pulmonary hypertension.

CTD, connective tissue disease; SSc, systemic sclerosis; 6MWD, 6-min walk distance; cGMP, cyclic guanosine monophosphate; NYHA, New York Heart Association; PVR, pulmonary vascular resistance; NT-pro-BNP, N-terminal pro-brain natriuretic peptide; PAP, pulmonary arterial pressure; CO, cardiac output; CI, cardiac index; SSc-PAH, systemic sclerosis with pulmonary arterial hypertension; RV, right ventricular.
Nitric oxide pathway
Phosphodiesterase type 5 (PDE-5) inhibitors, including sildenafil and tadalafil, enhance the effect of nitric oxide on pulmonary vascular smooth muscle cells, and are commonly used in patients with SSc-PAH, and have been shown to improve outcomes in patients with PAH (Table 5).54,55 Side effects of the PDE-5 inhibitors include headache, flushing, diarrhea and GI upset, but in general, this class of medications is well tolerated and orally administered, making it an appealing option for treatment.
Riociguat, a guanylate cyclase stimulator that increases sensitivity to nitric oxide, has recently been studied in PAH. The PATENT-1 (Pulmonary Arterial Hypertension Soluble Guanylate Cyclase-Stimulator Trial 1) study compared riociguat with placebo in patients with PAH (Table 5). 56 Common side effects in this study included headache, GI upset, peripheral edema and dizziness. There were also more syncopal events in subjects taking riociguat than in those taking placebo (4% versus 1%, respectively). The open-label extension PATENT-2 study demonstrated a similar side-effect profile and found that treatment with riociguat led to lasting improvement in 6-min walk distance (6MWD) and WHO functional class. 67
Endothelin pathway
Endothelin receptor antagonists (ERAs) are another appealing option for treatment of SSc-PAH. Endothelin-1 is produced by endothelial cells and has direct vasoconstrictive effects on pulmonary vascular smooth muscle cells. Bosentan, macitentan and ambrisentan have all been studied in PAH (Table 5).57–59 In an analysis of patients with connective tissue disease (CTD)-PAH in the ARIES-1 and ARIES-2 trials (ambrisentan), patients with CTD-PAH demonstrated improved 6MWD and fewer clinical worsening events compared with historical controls. 68 The SERAPHIN (Study with Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome) compared macitentan with placebo and was notable in that its primary endpoint was event driven rather than change in 6MWD. 58 The ACCP recommends first line treatment with either an ERA, PDE-5 inhibitor or riociguat for patients with WHO FC II or III PAH. 49
Prostacyclin pathway
The prostacyclin analogs act as vasodilators and play a role in the treatment of SSc-PAH (Table 5).60–63 However, given that many of the medications are intravenous or inhaled, administration can be challenging. The ACCP recommends use of a parenteral prostanoid for patients with FC III symptoms who have rapidly progressive disease or progression of disease despite treatment with one or two classes of oral agents, and for patients with FC IV symptoms. 49
The newest agent targeting the prostacyclin pathway is selexipag, an oral selective IP prostacyclin-receptor agonist (Table 5). In the GRIPHON (Prostacyclin (PGI2) Receptor Agonist in Pulmonary Arterial Hypertension) study, treatment of PAH patients with selexipag resulted in a 40% reduction in death from any cause or a complication related to PAH compared with a placebo group. 64
Combination therapies
Given the different pathways targeted by each of these medication classes, the idea of combination therapy upfront is appealing to provide synergistic or additive benefit in the treatment of PAH. The AMBITION (Ambrisentan and Tadalafil in Patients with Pulmonary Arterial Hypertension) study randomized 500 patients with PAH and WHO FC II or III symptoms to either a combination of ambrisentan plus tadalafil, ambrisentan plus placebo, or tadalafil plus placebo. 65 The primary endpoint was the time to a first event of clinical failure (including a composite of death, hospitalization for worsening PAH, disease progression, or unsatisfactory long-term clinical response), and an event occurred in 18% of the combination group compared with 31% of the pooled monotherapy group. A total of 37% of the patients had CTD-associated PAH. In a smaller study of 24 treatment-naïve patients with SSc-PAH, combination ambrisentan and tadalafil treatment resulted in reduction in RV mass and pulmonary vascular residence and an improvement in stroke volume, tricuspid annular plane systolic excursion, 6MWD, and serum NT-pro-BNP levels, suggesting that combination therapy may be of benefit in patients with SSc-PAH. 66
Anticoagulation
A number of retrospective and prospective studies in idiopathic PAH have suggested that anticoagulation may be associated with a survival benefit,69,70 although newer studies have not demonstrated an appreciable benefit. 71 There is less clear evidence with regard to the use of anticoagulation in SSc-PAH. One retrospective study using propensity-score matching found no clear benefit of warfarin in patients with SSc-PAH, 72 and another registry-based study found that warfarin use was associated with decreased survival in SSc-PAH. 71 On the other hand, a retrospective study of 117 patients with CTD-associated PAH, 95% of whom had SSc, demonstrated that warfarin use was associated with a reduction in mortality. 73 Given the variable results of these trials, routine anticoagulation in patients with SSc-PAH is not recommended at this time. To address the role of anticoagulation in SSc-PAH prospectively, the SPHInX (Scleroderma-Pulmonary Arterial Hypertension Intervention with Apixaban) trial, a double-blind randomized controlled trial, is currently in development.
Experimental therapies
Although current therapies do improve outcomes in patients with SSc-PAH, morbidity and mortality remain high. To that end, a number of studies are currently in development to address the use of alternative agents in SSc-PAH. A randomized controlled trial comparing rituximab, a CD20 monoclonal antibody, with placebo in SSc-PAH is currently recruiting patients and will measure change in pulmonary vascular resistance as its primary outcome measure [ClinicalTrials.gov identifier: NCT01086540]. Ifetroban, a thromboxane A2/prostaglandin-receptor antagonist, will be evaluated in an upcoming randomized, placebo-controlled, double-blind phase II trial of patients with either dcSSc or SSc-PAH. The primary outcome will measure the incidence of adverse events, but a number of other parameters will also be measured, including change in DLCO, ventricular function as determined by magnetic resonance imaging, and TTE and change in a number of blood and skin biomarkers [ClinicalTrials.gov identifier NCT0268251]. There is also interest in the role of statins in the treatment of some of the vascular complications of SSc, including PAH. Although no specific trials are planned currently, this is an area of active interest and investigation.
Summary
Both ILD and PAH remain a major cause of morbidity and mortality among patients with SSc. Screening and early detection of these comorbidities are important aspects in the care of patients with SSc and can help improve outcomes. As the mechanisms underlying the pathogenesis of SSc-ILD and SSc-PAH are further elucidated, more targeted treatment modalities have become available. As these new therapeutic options are trialed in larger studies, the hope is that they may provide more effective options for treatment of these two conditions.
Footnotes
Funding
This work was supported by funding from NIH T23-AR0072 and K08-AR062592.
Conflict of interest statement
The authors declare that there is no conflict of interest.