
Abstract
Background and aims:
Low peak inspiratory flow (PIF) is common following severe exacerbations of chronic obstructive pulmonary disease (COPD). Patients with COPD and low PIF may be at risk of suboptimal delivery of inhaled therapies to the airways, especially when using devices such as dry powder inhalers (DPIs), which require greater inspiratory effort than metered dose inhalers (MDIs). We report the results from a 2-week crossover study evaluating the effects of inhaled dual therapy with budesonide/formoterol fumarate dihydrate with an MDI with a spacer versus a DPI in patients with COPD and low PIF.
Methods:
This randomized, open-label, two-period (each 1 week in duration) crossover efficacy and safety study included patients with severe-to-very severe COPD and PIF < 50 L/min (NCT04078126). Patients were randomized 1:1 to twice-daily budesonide/formoterol fumarate dihydrate MDI (BFF MDI) 320/10 µg with a spacer for 1 week followed by twice-daily budesonide/formoterol fumarate dihydrate DPI (BUD/FORM DPI) 320/9 µg for 1 week, or the inverse. The primary endpoint was peak change from baseline in forced expiratory volume in 1 s (FEV1) within 4 h post-dose following 1 week of treatment. Other assessments included pre-dose lung function, pharmacokinetics, and safety, as assessed by adverse events.
Results:
The modified intention-to-treat analysis set comprised 30 patients (mean age: 66.9 years; mean baseline FEV1: 766 mL; mean COPD assessment test score: 22.20). Following 1 week of treatment, both BFF MDI and BUD/FORM DPI improved mean [95% confidence interval (CI)] peak FEV1 4 h post-dose [256 (190, 322) mL and 274 (208, 340) mL, respectively]. No clinically meaningful difference between treatments was observed for any lung function endpoint. There were no unexpected safety findings.
Conclusion:
Dual therapy with BFF MDI and with BUD/FORM DPI led to improvements in lung function in patients with severe-to-very severe COPD and low PIF.
Keywords
Introduction
Exacerbations of chronic obstructive pulmonary disease (COPD) are characterized by an acute worsening of respiratory symptoms and negatively impact the rates of hospitalization and readmission. 1 Low (i.e. ⩽ 60 L/min) peak inspiratory flow (PIF) has been reported in 31.7 2 –52% 3 of patients following severe exacerbations of COPD (i.e. exacerbations requiring hospitalization or a visit to the emergency room 1 ) and can result in a shorter time to hospital readmission. 3 Patients with COPD and low PIF may also be at risk of suboptimal delivery of inhaled medications to the airways, particularly when using devices with high inspiratory airflow resistance. 4
Importantly, the flow dependency of fine particle mass is greater for dry powder inhalers (DPIs) than metered dose inhalers (MDIs), which could impact medication delivery and subsequent lung function. 5 DPIs require the patient to produce sufficient airflow (typically ⩾ 60 L/min), 4 but their airflow resistance [0.060–0.163 cmH2O0.5 (L/min)−1] and the resulting PIF rate (49–108 L/min) can vary widely by device. 6 Although MDIs are less dependent on a patient’s ability to produce airflow than DPIs, some patients have difficulty appropriately using MDIs, including coordinating device activation and inhalation.6,7 To improve coordination of inhaler actuation and inspiration, a spacer (a valved holding chamber) may be used with an MDI. 7 Studies have shown that drug delivery may be improved using a spacer when MDI inhalation technique is suspected to be suboptimal.8,9 This may result in part from a reduced amount of drug being deposited in the oropharynx. 7
Fixed-dose inhaled corticosteroid (ICS)/long-acting β2-agonist (LABA) dual therapy is effective in improving lung function and reducing exacerbations in patients with moderate-to-very severe COPD and a history of exacerbations. 1 This study evaluated whether lung function outcomes would be differentially improved by delivering dual combination maintenance therapy with the ICS budesonide and the LABA formoterol fumarate dihydrate via an MDI with a spacer versus a DPI in patients with severe-to-very severe COPD and low PIF. Budesonide and formoterol fumarate steady-state pharmacokinetics were also measured to enable an assessment of exposure following administration with an MDI or DPI. In addition, safety was assessed, as measured by the occurrence of adverse events (AEs).
Methods
Study design
This was a phase IIIb, randomized, open-label, two-period (each 1 week in duration) crossover efficacy and safety pilot study (NCT04078126) conducted at four sites in Germany that compared the delivery of inhaled budesonide/formoterol fumarate dihydrate dual therapy with two different devices: twice-daily budesonide/formoterol fumarate dihydrate MDI 320/10 µg (BFF MDI) administered with a spacer versus twice-daily budesonide/formoterol fumarate dihydrate DPI 320/9 µg (BUD/FORM DPI; Symbicort® Turbuhaler®; AstraZeneca, Cambridge, UK). An overview of the study design is shown in Figure 1. Based on the previously published data, a 1-week treatment duration was considered sufficient to reach steady-state exposure of budesonide and formoterol fumarate dihydrate in both treatment arms 10 and to evaluate resulting increases in peak expiratory volume in 1 s (FEV1) within 4 h post-dose.11,12 The lung function and pharmacokinetic endpoints were objective measures not expected to be impacted by the open-label nature of the comparison, and the crossover design allowed within-patient comparisons to be made.

Study design.
This study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and that are consistent with International Council on Harmonisation/Good Clinical Practice and applicable regulatory requirements. The study protocol and its amendments, the informed consent form, and other relevant documents were reviewed and approved by an institutional review board/independent ethics committee before the study was initiated. Each patient or their legally authorized representative was required to provide written informed consent before participation.
This study was conducted between September 2019 and December 2020. On 17 March 2020, due to circumstances related to the coronavirus disease 2019 (COVID-19) pandemic, patients who were in screening or run-in, or who had been randomized but had not completed the study were considered screen failures and discontinued when the study was paused. On the study recommencing on 17 September 2020, all patients impacted by the study hold were reinvited and started with informed consent procedures before initiating study assessments as appropriate.
Patients
Patients eligible for study participation were current or former smokers (history of ⩾ 10 pack-years) aged 40–80 years (inclusive) who were diagnosed with COPD and receiving ⩾ 2 inhaled maintenance therapies for COPD in the 4 weeks before the first visit, including ⩾ 1 long-acting bronchodilator. All patients were required to demonstrate acceptable MDI with a spacer and DPI administration technique and to be able to correctly perform the PIF measurement.
At visit 2, eligible patients had a pre-bronchodilator PIF of < 50 L/min, as measured by an In-Check inspiratory flow measurement device (In-Check™ DIAL G16; Clement Clarke International, Harlow, UK) at Turbuhaler Symbicort (S) resistance; the In-Check device measures PIF in increments equal to 5 L/min and data were recorded to the nearest increment. Eligible patients also had a post-bronchodilator ratio of the FEV1 to forced vital capacity of < 0.70, and a post-bronchodilator FEV1 of < 50% of the predicted normal value. Because a PIF rate of 60 L/min is considered optimal for DPIs, we selected patients who had a PIF rate of ⩽ 50 L/min at Turbuhaler S resistance to evaluate for a clinically meaningful difference.
Key exclusion criteria included a current diagnosis of asthma, COPD due to α1-antitrypsin deficiency, sleep apnea, other respiratory disorders, or any other significant disease or disorder that could put the patient at risk or influence the results of the study. Patients were also ineligible if they had a moderate or severe exacerbation of COPD within 6 weeks before randomization, had lung resection or lung-volume-reduction surgery in the 6 months before the first visit, or required mechanical ventilation in the 3 months before visit 1.
Procedures and treatments
At visit 1, eligible patients discontinued their COPD maintenance therapies and entered a 2- to 3-week run-in period receiving ipratropium bromide/fenoterol hydrobromide 20/50 µg (Berodual® Respimat®; Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany) four times daily. Patients receiving ICSs at screening also received budesonide MDI 320 µg twice daily. Albuterol sulfate [Ventolin® hydrofluoroalkane (HFA); GlaxoSmithKline, Brentford, UK] was provided as rescue therapy throughout the study and could be used as needed; however, rescue therapy was withheld for at least 6 h prior to spirometry measurements on study visit days. At visit 2, PIF was measured with the In-Check inspiratory flow measurement device, set to no resistance, Turbuhaler S resistance, and Ellipta device (GlaxoSmithKline, Brentford, UK) resistance; reversibility and COPD assessment test (CAT) scores were also measured.
Following the run-in period, patients who remained eligible and who had baseline FEV1 stability at visit 3 (i.e. within 20% or 200 mL of the pre-bronchodilator assessment at visit 2) discontinued run-in treatments and were randomized 1:1 to one of the two treatment sequences: open-label BFF MDI 320/10 µg administered with a spacer twice daily for 1 week followed by a 2-week washout period, then followed by open-label BUD/FORM DPI 320/9 µg administered twice daily for 1 week, or the inverse (Figure 1). During the 2-week washout period, patients discontinued their randomized treatment and resumed their run-in treatments. Randomization was stratified by PIF values at visit 3 (< 40 versus ⩾ 40 L/min at Turbuhaler S resistance).
Patients were admitted to the clinic prior to the evening doses of study treatment on the days before visits 3, 4, 5, and 6; pre- and post-dose spirometry assessments were conducted at each of these visits. Ipratropium bromide/fenoterol hydrobromide and budesonide were withheld after evening dosing before visits 3 and 5 to allow for a washout period of at least 12 h prior to spirometry on the visit day. At visits 4 and 6, the time of dosing was standardized to be > 11 h and < 12 h from the previous evening dose so as to remove any confounding based on prior doses.
Samples of approximately 10 mL of whole blood were drawn for pharmacokinetic analyses 30 min pre-dose and 2, 5, 20, 30, 40, 60, 120, 180, and 240 min post-dose on the morning of visits 4 and 6. Budesonide and formoterol plasma concentrations were determined by Covance, Inc. (Salt Lake City, UT, USA), as previously described. 13
Patients were considered to have completed the study when they had finished the last scheduled procedure and the follow-up telephone call, which was scheduled to take place 7–14 days after the last dose.
Endpoints
The primary endpoint was the peak change from baseline in FEV1 within 4 h following 1 week of treatment. Secondary efficacy endpoints following the first dose of each treatment on day 1 were change from baseline in 2-h FEV1 and change from baseline in 2-h inspiratory capacity (IC). Additional secondary efficacy endpoints following 1 week of treatment were the area under the curve for change from baseline in FEV1 from 0 to 4 h (FEV1 AUC0–4), change from baseline in pre-dose FEV1, change from baseline in 2-h post-dose IC, and change from baseline in pre-dose PIF (In-Check device set to no resistance, Turbuhaler S resistance, or Ellipta resistance).
Key pharmacokinetic endpoints for budesonide and formoterol fumarate included area under the concentration–time curve from 0 to 4 h post-dose (AUC0–4), maximum observed plasma concentration (Cmax), time to Cmax (tmax), and concentration at the end of the dosing interval (Ctrough).
Safety endpoints were AEs, serious AEs, and AEs leading to treatment discontinuation.
Statistical analysis
It was planned for approximately 30 patients to be randomized (15 per treatment sequence) for 26 patients to complete the study. Sample size was selected based on practical considerations to obtain reasonable point estimates of treatment effects related to each device.
No formal hypothesis testing was conducted in this pilot study; only exploratory hypotheses were evaluated, and there were no corrections for multiplicity. Statistical analyses were conducted using SAS® version 9.4 (SAS Institute, Cary, NC).
The intention-to-treat (ITT) analysis set comprised all randomized patients who received ⩾ 1 dose of study treatment.
To isolate differences due to device only, a per-protocol estimand was used, defined as the effect of the randomized treatments in all patients who were compliant with the protocol and not impacted by other factors, such as adherence, technique, or dosing time. Thus, analyses for the per-protocol estimand used the modified ITT (mITT) analysis set that included patients in the ITT analysis set who had post-baseline spirometry data from both treatments at visits 4 and 6 and no significant protocol violations. Patients who did not restart the study after it was paused due to the COVID-19 pandemic were included in the ITT analysis set but not in the mITT analysis set; those who restarted the study were included in the ITT or mITT analysis set only for the instance of enrollment after restarting, providing they met the definition for the analysis set as described.
Efficacy analysis
The primary endpoint was analyzed using an analysis of covariance (ANCOVA) model with baseline FEV1, PIF at screening (at Turbuhaler S resistance), and reversibility to albuterol sulfate HFA as continuous covariates; treatment and period were included as categorical covariates. The model included patient as a random effect and did not include patient sequence unless it was determined to be statistically important (p < 0.1). Estimates of the difference between treatments with 95% confidence intervals (CIs) are reported with p values provided to aid interpretation. Secondary efficacy endpoints in the mITT analysis set and peak change from baseline FEV1 4 h post-dose in the ITT analysis set were analyzed using a similar model.
The primary and secondary endpoints were also analyzed in subgroups based on PIF (< 40 and ⩾ 40 L/min) at screening based on Turbuhaler S resistance. Scatterplots and Pearson’s correlation coefficients were used to investigate associations between baseline PIF on a continuous scale and within-patient differences in device effect for the primary endpoint.
Pharmacokinetics analysis
Pharmacokinetics were analyzed in the pharmacokinetics analysis set, which included all patients in the mITT analysis set with Cmax defined in both treatment periods and no significant protocol violations that influenced the pharmacokinetic analyses; patients were analyzed according to treatment received.
Pharmacokinetic parameters were derived using noncompartmental methods in Phoenix® WinNonlin® version 8.1 or higher software (Certara, Inc., Princeton, NJ, USA). Cmax, Ctrough, and tmax were obtained from the concentration–time profiles, with the concentration at the end of the dosing interval defined as the pre-dose concentration at visits 4 and 6. Logarithmic transformations of Ctrough, Cmax, and AUC0–4 were analyzed with an ANCOVA model, using treatment and period as categorical covariates and patient as a random effect; treatment sequence was only included in the model if determined to be statistically important (p < 0.10). Ratios and 90% CIs for Cmax, Ctrough, and AUC0–4 were generated and produced by back-transformation. Descriptive statistics for untransformed tmax are presented.
Safety analysis
Safety was analyzed in the safety analysis set, which comprised patients who were randomized and received at least one dose of study treatment; patients were analyzed by the treatment received. Patients who restarted the study after it was paused were included in the safety analysis set only for the instance of enrollment after restarting. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 23.1.
Results
Patient disposition
The first patient was enrolled on 10 September 2019; the last patient had their last visit on 30 December, 2020. Patient disposition is summarized in Figure 2. In brief, 77 patients were enrolled and 35 were randomized to receive treatment (n = 18 to receive BFF MDI followed by BUD/FORM DPI; n = 17 to receive BUD/FORM DPI followed by BFF MDI). As it is difficult to predict a low PIF in a patient based on other clinical characteristics or lung function measurements, a relatively high number of patients needed to be screened to randomize this study. Overall, 30/35 (85.7%) of randomized patients were included in the mITT population. The pharmacokinetic analysis set included 28 patients. Four patients [4/35 (11.4%)] discontinued the study early due to the COVID-19 pandemic-related study hold.
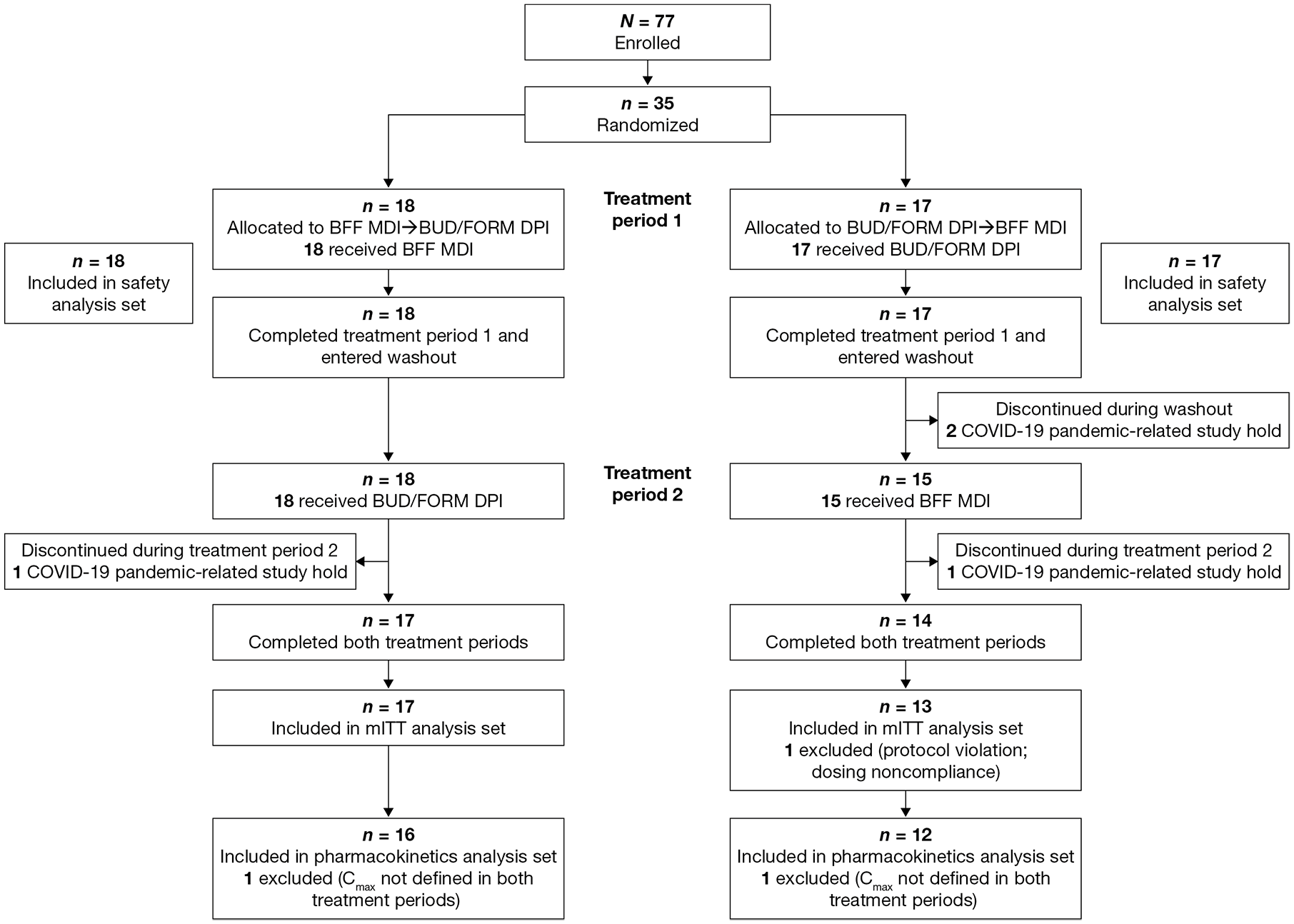
Patient disposition.
Demographics and baseline characteristics
Patient demographics and baseline clinical characteristics in the mITT analysis set are reported in Table 1. In brief, all patients were White and not of Hispanic or Latino ethnicity; 50% of the patients were female; and the mean [standard deviation (SD)] age was 66.9 (4.8) years. Overall, the median time since diagnosis with COPD was 11.7 (minimum 2.8, maximum 30.7) years; the majority of patients had severe COPD and the mean (SD) post-bronchodilator FEV1 was 32.52% (8.15%) of the predicted normal value. The mean (SD) FEV1 at baseline was 766 (212) mL, and the mean (SD) CAT score was 22.20 (6.13). Approximately half [14/30 (46.7%)] of the patients had ⩾ 1 COPD exacerbation in 12 months before the study. Mean (SD) baseline PIF values with the In-Check Device set to no resistance, Turbuhaler S resistance, and Ellipta resistance were 78.86 (25.81), 38.56 (9.60), and 47.53 (13.60) L/min, respectively.
Patient demographics and baseline COPD characteristics (mITT analysis set).

CAT, COPD assessment test; COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 s; FEV1% predicted, FEV1 percentage of the predicted normal value; GOLD, Global Initiative for Chronic Obstructive Lung Disease; mITT, modified intention-to-treat; PIF, peak inspiratory flow; SD, standard deviation; Turbuhaler S, Turbuhaler Symbicort.
Relative to day 1 of the first treatment period.
Defined as the mean of the last pre-dose values at visits 3 and 5. If both values were missing, the last pre-dose assessment during screening visits was used.
Efficacy
Similar improvements in peak FEV1 within 4 h post-dose following 1 week of treatment were observed for both BFF MDI and BUD/FORM DPI (Figure 3). Adjusted mean [95% confidence interval (CI)] changes from baseline in peak FEV1 were 256 (190, 322) mL for BFF MDI and 274 (208, 340) mL for BUD/FORM DPI, corresponding to a between-group difference of −17 (−54, 20) mL (p = 0.3450). Similarly, the adjusted mean difference (95% CI) in the ITT analysis set was −16 (−51, 19) mL. Patients with PIF ⩾ 40 L/min at screening (n = 18) had numerically greater improvements in peak FEV1 within 4 h post-dose following 1 week of treatment compared with those with PIF < 40 L/min (n = 12) for both BFF MDI and BUD/FORM DPI (Supplemental Table S1). Between-treatment differences for changes in peak FEV1 within 4 h post-dose in individual patients were not correlated with baseline PIF at any resistance level tested (Supplemental Figure S1).

Peak change from baseline in FEV1 within 4 h post-dose following 1 week of treatment (mITT analysis set).
Following the first dose, improvements in 2-h post-dose FEV1 were numerically greater with BFF MDI compared with BUD/FORM DPI, but improvements in 2-h post-dose IC were similar between treatments (Table 2). After 1 week of treatment, improvements of similar magnitude were observed for FEV1 AUC0–4, pre-dose FEV1, and 2-h post-dose IC with both BFF MDI and BUD/FORM DPI (Table 2). Improvements in secondary efficacy endpoints were generally larger in patients with PIF ⩾ 40 L/min at screening compared with those with PIF < 40 L/min for both BFF MDI and BUD/FORM DPI (Supplemental Table S1).
Secondary efficacy endpoints (mITT analysis set).
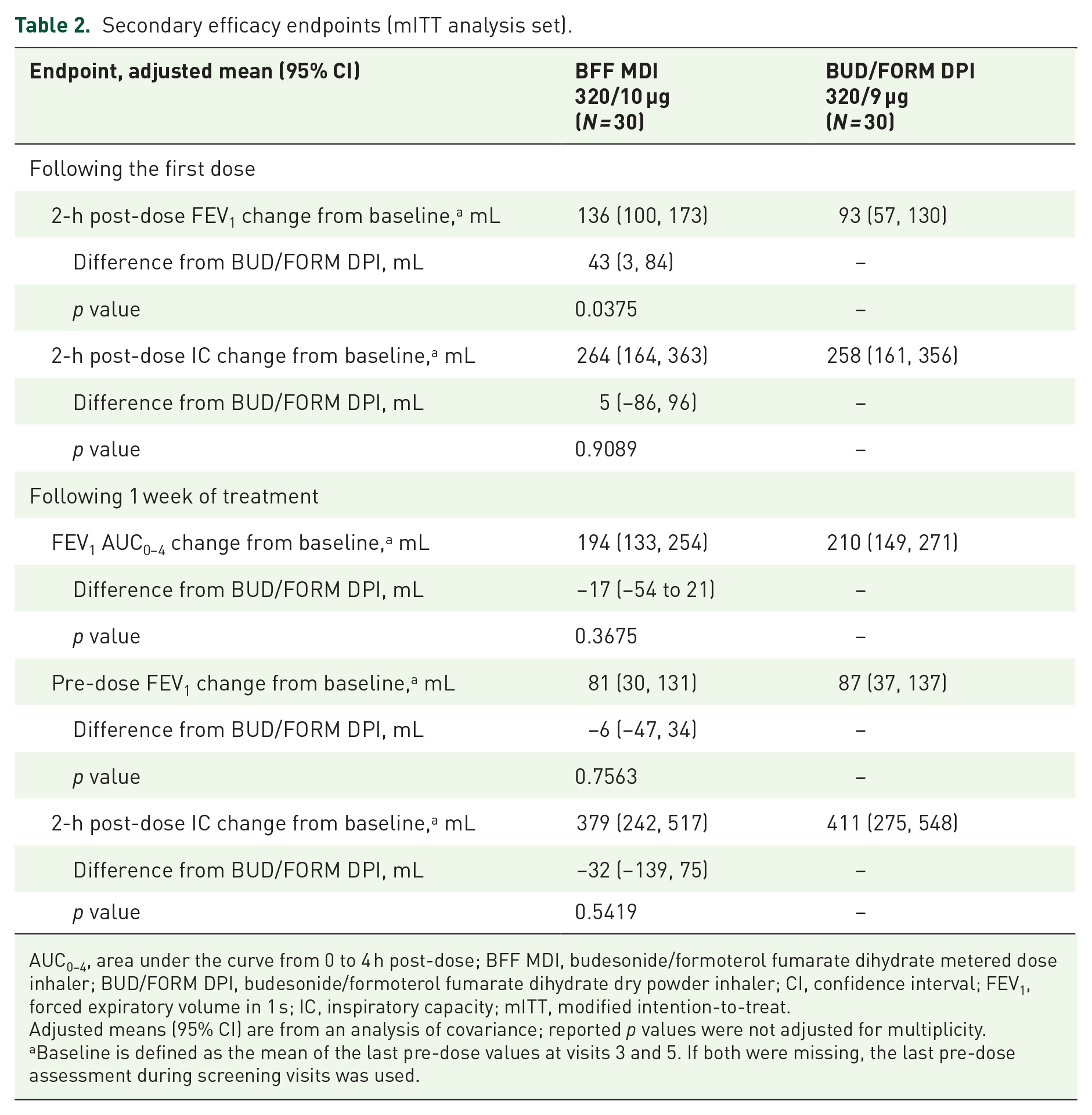
AUC0–4, area under the curve from 0 to 4 h post-dose; BFF MDI, budesonide/formoterol fumarate dihydrate metered dose inhaler; BUD/FORM DPI, budesonide/formoterol fumarate dihydrate dry powder inhaler; CI, confidence interval; FEV1, forced expiratory volume in 1 s; IC, inspiratory capacity; mITT, modified intention-to-treat.
Adjusted means (95% CI) are from an analysis of covariance; reported p values were not adjusted for multiplicity.
Baseline is defined as the mean of the last pre-dose values at visits 3 and 5. If both were missing, the last pre-dose assessment during screening visits was used.
Across the resistances tested, improvements in pre-dose PIF were generally less than one increment on the measurement scale (5 L/min) on the In-Check device for both BUD/FORM DPI and BFF MDI, irrespective of PIF at screening, and were not considered clinically meaningful (Supplemental Table S2).
Pharmacokinetics
Plasma concentration–time profiles for budesonide were similar between BFF MDI and BUD/FORM DPI (Figure 4(a)). The rate and extent of budesonide absorption between BFF MDI and BUD/FORM DPI were similar based on median tmax and geometric mean Cmax, AUC0–4, and Ctrough (Table 3); however, there was high between-patient variability in budesonide exposure with both treatments. Adjusted geometric mean treatment ratios for AUC0–4 and Cmax were approximately 1, with the 90% CI falling within the range of 0.8–1.25 (Table 3). For Ctrough, the adjusted geometric mean ratio for was < 1 with a wide 90% CI, indicating that budesonide trough concentrations were lower with BFF MDI compared with BUD/FORM DPI (Table 3).
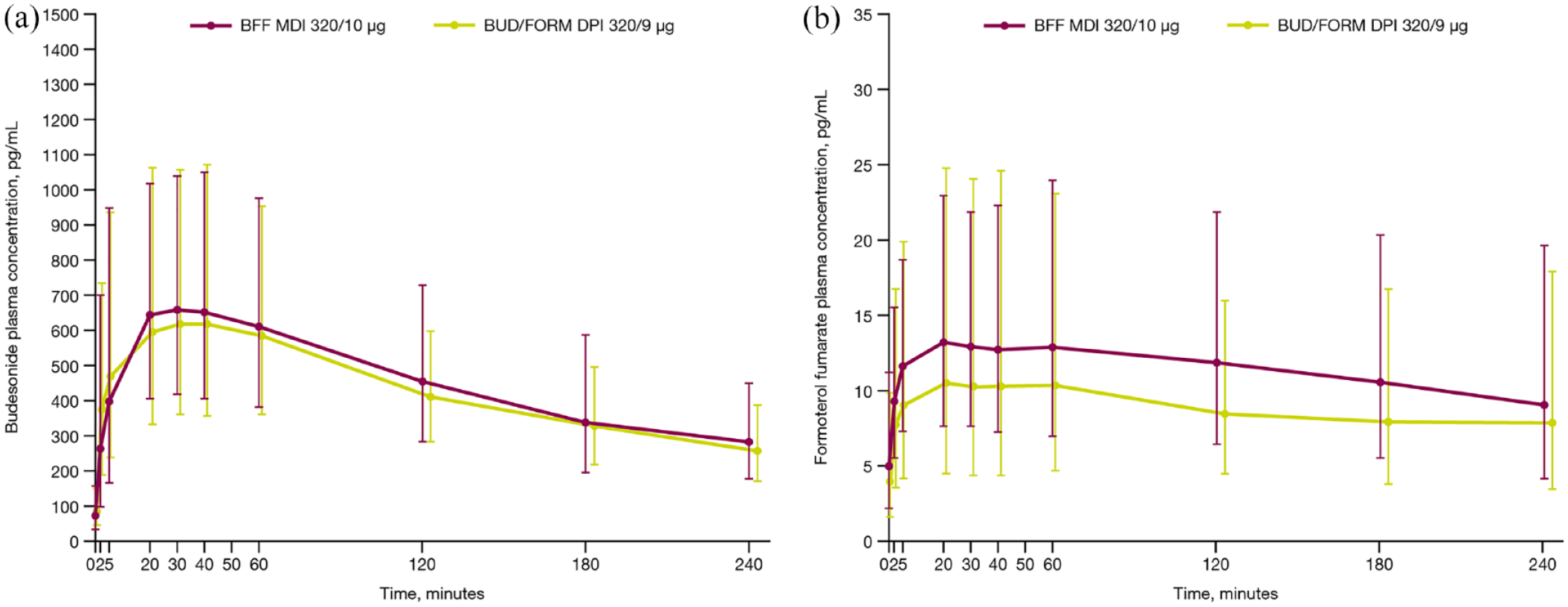
Geometric mean (a) budesonide and (b) formoterol fumarate plasma concentrations over time (pharmacokinetics analysis set).BFF MDI, budesonide/formoterol fumarate dihydrate metered dose inhaler; BUD/FORM DPI, budesonide/formoterol fumarate dihydrate dry powder inhaler.
Budesonide and formoterol fumarate pharmacokinetic parameters (pharmacokinetics analysis set).
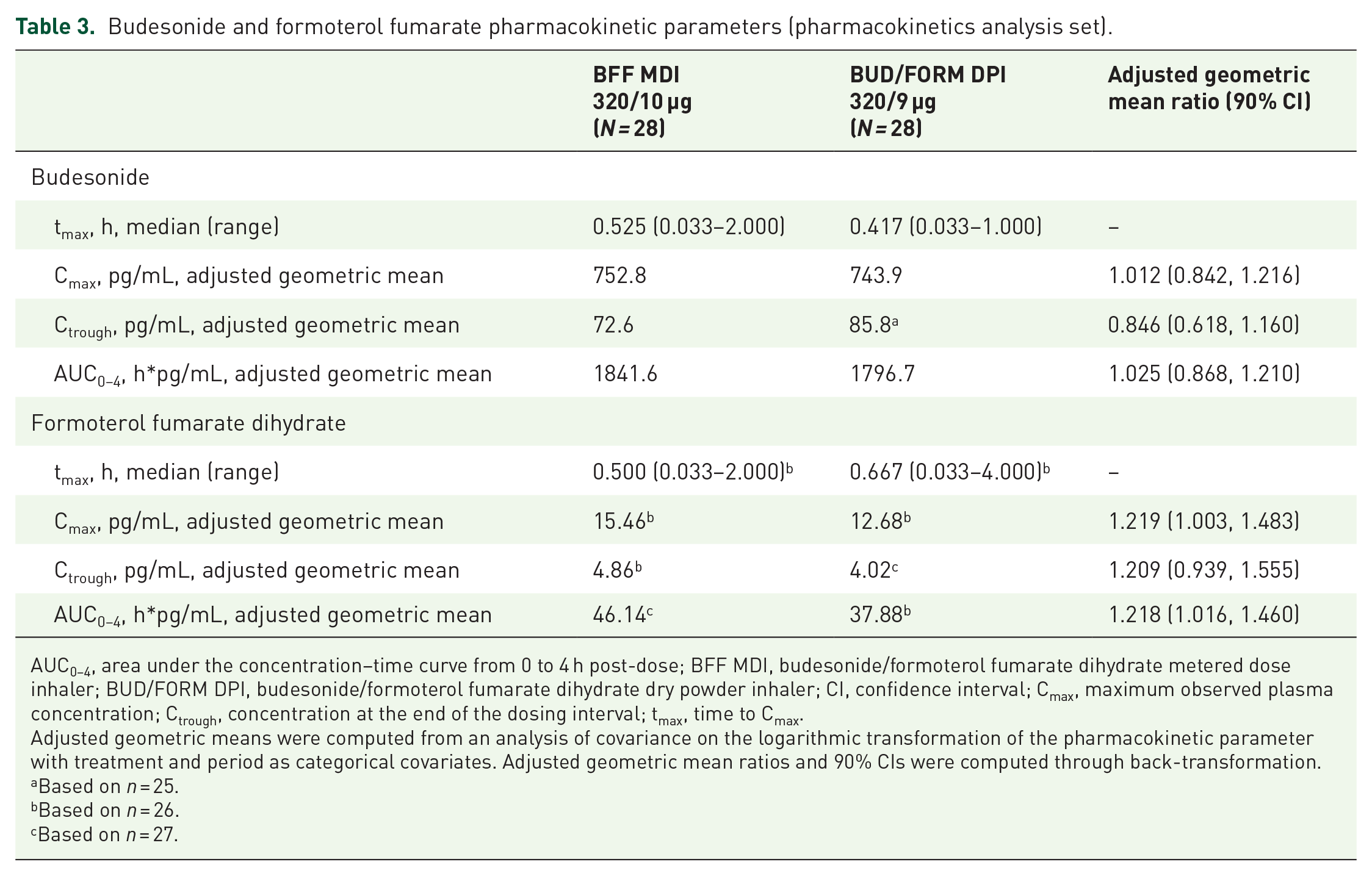
AUC0–4, area under the concentration–time curve from 0 to 4 h post-dose; BFF MDI, budesonide/formoterol fumarate dihydrate metered dose inhaler; BUD/FORM DPI, budesonide/formoterol fumarate dihydrate dry powder inhaler; CI, confidence interval; Cmax, maximum observed plasma concentration; Ctrough, concentration at the end of the dosing interval; tmax, time to Cmax.
Adjusted geometric means were computed from an analysis of covariance on the logarithmic transformation of the pharmacokinetic parameter with treatment and period as categorical covariates. Adjusted geometric mean ratios and 90% CIs were computed through back-transformation.
Based on n = 25.
Based on n = 26.
Based on n = 27.
For formoterol fumarate, plasma concentration–time profiles were slightly higher with BFF MDI with a spacer versus BUD/FORM DPI, although standard deviation estimates overlapped at all time points (Figure 4(b)). Despite formoterol fumarate absorption rates being similar across treatments, as evidenced by comparable median tmax values, the extent of absorption was greater with BFF MDI based on geometric mean treatment ratios for AUC0–4 (26.3% higher with BFF MDI), Cmax (24.5% higher with BFF MDI), and Ctrough values (18.5% higher with BFF MDI; Table 3). As with budesonide, between-patient variability in formoterol fumarate exposure was high with both treatments. Adjusted geometric mean treatment ratios from the overall statistical analysis exceeded 1 for AUC0–4, Cmax, and Ctrough, indicating that exposure to formoterol fumarate was higher with BFF MDI than BUD/FORM DPI (Table 3).
Safety
Two patients, one in each treatment arm, had a single treatment-emergent AE during the study. One patient had a moderate nonserious AE of angina, and one had a mild nonserious AE of abdominal discomfort. Both AEs were considered unrelated to study treatment by the investigator, and patients recovered with no changes to the administration of study treatment.
There were no deaths, no serious AEs, no AEs considered related to the study drug, and no AEs leading to discontinuation of the study drug during the study.
Discussion
In this phase IIIb pilot study, treatment with either BFF MDI or BUD/FORM DPI for 1 week was associated with clinically meaningful improvements compared with baseline in peak FEV1 within 4 h post-dose in patients with severe-to-very severe COPD and low PIF (< 50 L/min at Turbuhaler S resistance). Notably, these improvements in FEV1 of > 250 mL were observed in a study population with markedly limited lung function, as evidenced by the mean baseline FEV1 of 766 mL. In subgroups defined by PIF (<40 or ⩾40 L/min) at screening, improvements in FEV1 were also similar across treatments. In addition, correlational analyses showed that there was no association between baseline PIF and the differences in FEV1 improvements between treatments. Both treatments resulted in improved lung function, but a clinically meaningful difference was not observed in peak change from baseline in FEV1 between BFF MDI and BUD/FORM DPI.
Given the high inspiratory airflow resistance offered by DPIs compared with MDIs, it was hypothesized that the delivery of maintenance medication via an MDI with a spacer would be more effective in patients with low PIF than delivery via a DPI, thereby resulting in greater improvements in lung function with the use of an MDI. The current findings do not support this hypothesis, and both devices were effective in this patient population. Improvements were also observed for secondary lung function endpoints with each treatment. As for the primary endpoint, there were generally no clinically meaningful differences between BFF MDI and BUD/FORM DPI. Although a numerically greater improvement in 2-h post-dose FEV1 was observed following the first dose of treatment with BFF MDI versus BUD/FORM DPI, this finding was not sustained following 1 week of treatment and is not likely to be clinically meaningful. Similarly, both the absolute changes from baseline in pre-dose PIF and differences observed between BFF MDI and BUD/FORM DPI were small and not considered clinically meaningful across all tested resistances.
Improvements in lung function were generally numerically greater across endpoints in patients with PIF at screening ⩾ 40 L/min versus those with PIF < 40 L/min. However, this was observed for both treatments, and the differences were not clinically meaningful in either subgroup for any endpoint. Furthermore, correlational analysis of the difference between the treatments for the primary endpoint and PIF at screening revealed no association. Taken together, there was no evidence to suggest that the conclusions of the study would have differed, had a lower PIF threshold been used for inclusion in the study.
The rate of absorption of budesonide and formoterol fumarate was generally similar between BFF MDI and BUD/FORM DPI, with similar tmax observed for both treatments with each device. Although the extent of budesonide exposure was similar with BFF MDI and BUD/FORM DPI, formoterol fumarate exposure was slightly higher with BFF MDI compared with BUD/FORM DPI. The higher formoterol fumarate exposure observed with BFF MDI may be partially attributed to the higher dose administered versus BUD/FORM DPI (10 versus 9 µg, respectively) and is consistent with at least one previously published study. 13 However, the roughly 11% higher dose may not account for the entire 22% difference in Cmax and AUC0–4. Differences between the devices may potentially contribute, although the small sample size and high variability between patients limit the ability to determine if the difference in formoterol fumarate exposure is related to delivery device. In either case, this difference was not associated with any meaningful difference with respect to lung function by week 1.
Despite the study being interrupted by the COVID-19 pandemic, there were no apparent issues related to the COVID-19 pandemic that impacted on the evaluation of efficacy in the study.
Both BFF MDI and BUD/FORM DPI were well tolerated. Only two AEs were reported in the study, and neither impacted the administration of study treatment. No AEs were known to be related to COVID-19. Although the small number of events precludes drawing firm conclusions, there were no unexpected safety findings.
Limitations of this pilot study include its small sample size and short treatment duration. It is also unclear whether the study results would have differed if the recruited population had low PIF due to an immediately preceding exacerbation rather than recruitment of patients with naturally low PIF away from the time of an exacerbation (<50% of this study’s population had an exacerbation in the last year, and having a moderate or severe exacerbation within 6 weeks of randomization was exclusionary). Although the baseline mean PIF values at Turbuhaler S and Ellipta resistances in the current population (38.56 and 47.53 L/min) approximated suboptimal PIF levels observed in hospitalized patients with COPD,3,14 low PIF in hospitalized patients is also associated with low grip strength 14 (a measure of general muscle weakness 15 ), which could impact the patient’s ability to effectively use their inhaler device. Thus, it is possible that in a peri-hospitalized population with low PIF the findings could have differed and that benefits of the use of an MDI with a spacer might have been observed.
Conclusion
In conclusion, both BFF MDI and BUD/FORM DPI improved lung function in patients with severe-to-very severe COPD and low PIF, without differential effects being observed based on the delivery device.
Supplemental Material
sj-docx-1-tar-10.1177_17534666221107312 – Supplemental material for Effect of inhaled budesonide/formoterol fumarate dihydrate delivered via two different devices on lung function in patients with COPD and low peak inspiratory flow
Supplemental material, sj-docx-1-tar-10.1177_17534666221107312 for Effect of inhaled budesonide/formoterol fumarate dihydrate delivered via two different devices on lung function in patients with COPD and low peak inspiratory flow by Bärbel Huber, Claus Keller, Martin Jenkins, Abid Raza and Magnus Aurivillius in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The authors would like to thank Debasree Purkayastha (for assistance with statistical analyses), Sara Asimus (for assistance with pharmacokinetic analyses), Wolfgang Timmer (for study investigation and oversight), Martin Hoffman (for study investigation), Heiner Steffen (for study investigation), Patrick Darken (for study conceptualization and interpretation), and Paul Dorinsky (for study conceptualization and interpretation) for their contributions to this study.
Ethics approval and consent to participate
This study was performed in accordance with ethical principles having their origin in the Declaration of Helsinki and that are consistent with International Council on Harmonisation/Good Clinical Practice and applicable regulatory requirements. The study protocol and its amendments, the informed consent form, and other relevant documents were reviewed and approved by an institutional review board/independent ethics committee before the study was initiated. Each patient or their legally authorized representative was required to provide written informed consent before participation.
Consent for publication
As part of the informed consent process, patients consented to publication of these data.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by AstraZeneca. Medical writing support, under the guidance of the authors, was provided by Brian Woolums, PhD of CMC Connect, McCann Health Medical Communications, and was funded by AstraZeneca, in accordance with Good Publication Practice (GPP3) guidelines (Battisti WP, Wager E, Baltzer L, et al. Ann Intern Med 2015;163: 461–464).
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: B.H. is an employee of Inamed Gmbh (part of Nuvisan Gmbh), which was contracted by AstraZeneca for this study. C.K. has a lung and bronchial medicine practice, including a study center in Frankfurt am Main, Germany and was contracted by AstraZeneca as a consultant research physician for this study. A.R. is an employee of Medsearch UK Limited and was contracted by AstraZeneca as a consultant research physician for this study. M.A. and M.J. are employees of AstraZeneca and own stock or stock options in the company.
Availability of data and materials
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.