
Abstract
Background:
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal interstitial lung disease (ILD). Currently, two antifibrotic drugs are available for reducing forced vital capacity (FVC) decline in IPF. However, many pulmonologists wait before initiating treatment, especially when IPF patients have stable disease. This study aimed to investigate the impact on survival outcome of FVC decline and a slow rate of FVC decline prior to and following treatment with these two antifibrotic drugs.
Methods:
Out of the 235 IPF patients treated with antifibrotic therapy that were screened, 105 cases were eligible, who then underwent physiological evaluation at 6 months prior to and following antifibrotic therapy. Clinical characteristics and prognostic outcomes were compared among groups, and prognostic factors were evaluated using a Cox proportional hazards analysis.
Results:
In terms of %FVC decline prior to the therapy and a slow rate of FVC decline, there was no significant difference between stable and worsened groups and responder and non-responder groups, respectively. On the other hand, in terms of %FVC decline (decline >5%) following antifibrotic therapy, the stable/improved group had significantly better prognosis than the worsened group. Prognostic analysis revealed that a stable/improved status following antifibrotic therapy [HR: 0.35 (0.15–0.87)] was significantly associated with a better prognosis.
Conclusions:
Concerning the FVC decline prior to and following antifibrotic therapy and a slow rate of FVC decline, only the FVC decline following the therapy is associated with a greater survival outcome. An early treatment decision may thus be beneficial for IPF.
The reviews of this paper are available via the supplemental material section.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and ultimately fatal interstitial lung disease (ILD) characterized by radiologic and/or histopathologic findings of usual interstitial pneumonia.1–3 As the disease progresses, lung function declines, which is accompanied by worsening of dyspnea and functional capacity. Acute exacerbations (AE) of IPF can occur in the course of the disease, and are associated with high mortality. 4 The majority of patients with IPF die from AE or respiratory failure.
Currently, two antifibrotic drugs, pirfenidone (PFD) and nintedanib (NTD), are available for reducing forced vital capacity (FVC) decline.5,6 The efficacy of these drugs has also been confirmed in real-world studies.7–10 However, many pulmonologists wait before initiating treatment, especially when IPF patients have stable disease.
We recently reported on an early marginal decline in FVC following treatment with PFD, which has a significant prognostic impact on IPF patients. 11 However, the impacts on survival of FVC decline prior to treatment and a slow rate of decline of FVC is not known. Furthermore, differences in the impact on survival of FVC decline following antifibrotic therapy with PFD or NTD have not also been studied.
Here, we investigate the impact on survival outcome of the disease behavior based on FVC and a slow rate of FVC decline prior to and following treatment with these two antifibrotic drugs.
Patients and methods
Patients and diagnostic criteria
We retrospectively included 235 patients with IPF who were treated with either PFD or NTD at Hamamatsu University Hospital and its related hospitals from 2009 to 2018. The diagnosis of IPF was based on the international consensus criteria of a combination of high-resolution computed tomography (HRCT) and surgical lung biopsy (SLB) findings. 1 The criteria for AE of IPF were in accordance with proposed international working group report of 2016. 4 To investigate the relationship between prognosis and the transition of FVC following treatment with antifibrotic therapy, we extracted cases that could be confirmed by pulmonary function tests 6 months prior to and following treatment. Cases with insufficient clinical information were defined as those where treatment was taken for less than 6 months, or where antifibrotic therapy was initiated after AE were excluded. Those who underwent lobectomy for lung cancer during the observation period were also excluded. Finally, a total of 105 patients were enrolled in the present study (Figure 1). The study protocol was approved by the ethics committees of Hamamatsu University School of Medicine (No 18-198) and all other hospitals. Patient approval, or the requirement for informed consent, was waived because of the retrospective nature of the review.

Study flow diagram. We conducted a retrospective review of 235 IPF patients treated with antifibrotic therapy. Finally, 105 patients were enrolled in the study, following the exclusion criteria listed.
Data collection
Clinical data such as age, sex, smoking status, body mass index (BMI), treatment for IPF, adverse events, and outcome were extracted from patients’ medical records. Laboratory and pulmonary function test findings were collected as well.
Physiological assessment prior to and following antifibrotic therapy
The %FVC was collected for 6 months prior to and following the start of treatment, and those whose %FVC (delta % FVCfollowing 6m) decreased by 5% or more after 6 months of starting treatment, were defined as the worsened group. On the other hand, patients whose %FVC decreases were less than 5% were defined as the stable/improved group. Similarly, patients with a %FVC decrease of 5% or more over the 6-month period prior to the start of treatment (delta%FVCprior to 6m) was defined as the worsened group.
We also investigated the slow rate of FVC decline prior to and following antifibrotic therapy. Non-responders were defined as those whose %FVC 6 months after starting treatment were lower than the values calculated from the rate of decline prior to treatment. Conversely, the group that showed higher %FVC after treatment than the expected value from the pre-treatment course were classified as responders (Figure S1).
Statistical methods
All values were expressed as medians (range) or numbers (%). The chi-squared or Mann–Whitney U tests were used for two-group comparisons. The cumulative survival and AE probabilities were evaluated using the Kaplan–Meier method, and the log-rank or Gray test was performed. Patients were censored if they remained alive until 31 December 2019. Cox proportional and Fine-Gray hazards analyses and multiple logistic regression analysis were used to identify significant variables that could predict survival status and AE, respectively. All statistical analyses were performed using EZR software version 1.36 (Saitama Medical Center, Jichi Medical University, Saitama, Japan). A p value of 0.05 was considered significant.
Results
Patient characteristics
The baseline characteristics of 105 IPF patients treated with PFD or NTD are summarized in Table 1. There were 59 cases treated with PFD and 46 cases treated with NTD. The median age was 70 years for both groups. The medians of BMI, PaO2, KL-6, %FVC, and %DLco (diffusion capacity for carbon monoxide) were 23.0 kg/m2 and 22.9 kg/m2, 79.4 Torr and 72.3 Torr, 1005 U/ml and 1118 U/ml, 67.5% and 68.1%, and 51.7% and 54.0% for the PFD and NTD groups, respectively. During the observation period, three patients in both groups discontinued treatment due to adverse events. Five patients in the PFD group and two patients in the NTD group initiated steroid therapy prior to antifibrotic therapy.
Patient characteristics.

Categorical data are presented as numbers (percentages) or medians (range).
BMI, body mass index; DLco, diffusion capacity for carbon monoxide; FVC, forced vital capacity; KL-6, Krebs von den Lungen 6; NTD, nintedanib; PaO2, partial pressure of oxygen; PFD, pirfenidone; SP-D, surfactant protein-D.
% FVC decreased by less than 5% 6 months after treatment; worsened, % FVC decreased by 5% or more at 6 months after treatment.
Relationship between %FVC decline and prognostic outcomes
The survival curves of the whole group, the PFD group, and the NTD group are shown in Figure 2. They were classified into stable or worsened groups based on their %FVC 6 months prior to treatment, respectively. There were no differences in survival curves between stable and worsened groups. Figure 3 shows the survival curves of the differences in the slow rate of FVC decline, which was examined with the criteria of whether or not the patient was a “responder”. There were no significant differences observed among the whole group, the PFD group, and the NTD group. The survival curves of patients with IPF defined by % FVC changes following antifibrotic therapy are shown in Figure 4. In the whole group and patients treated with NTD, the stable/improved groups for each showed significantly longer survival [HR; 0.36 (p < 0.01), 0.14 (p < 0.01), respectively]. In patients treated with PFD, the stable/improved group tended to have increased survival (p = 0.06).

Kaplan–Meier plot of survival rates as defined by %FVC 6 months prior to antifibrotic therapy and grouped by “stable” or “worsened” status. We examined whether %FVC decline prior to the start of treatment was related to prognosis. There was no significant difference between the prognosis of the “stable” group and “worsened” group. (a) All patients. (b) PFD group. (c) NTD group.
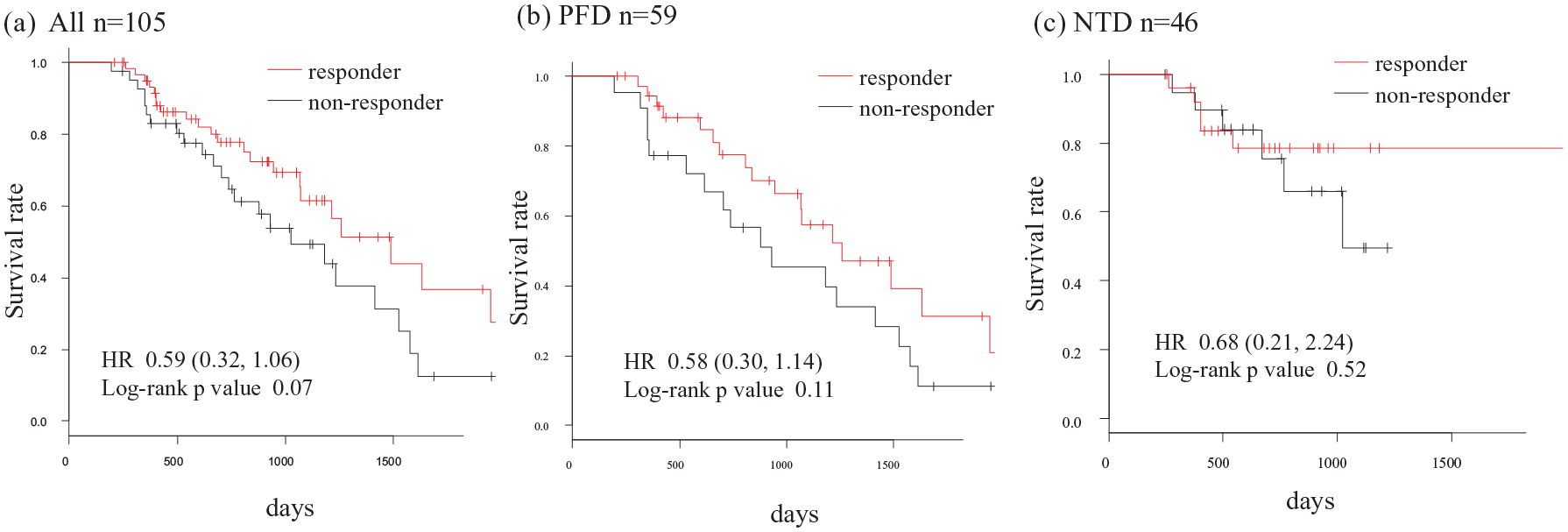
Kaplan–Meier plot of survival rates as defined by the differences in the slow rate of FVC decline prior to and following antifibrotic therapy grouped by “responder” or “non-responder” status. There was no significant difference between the prognosis of the “responder” and “non-responder” group. (a) All patients. (b) PFD group. (c) NTD group.

Kaplan–Meier plot of survival rate defined by %FVC 6 months following antifibrotic therapy grouped by “stable/improved” or “worsened” status. (a) All patients. The stable/improved group had a significantly better prognosis compared with the worsened group as classified by a 5% change in %FVC following 6 months of antifibrotic therapy in patients with IPF (log-rank test; p < 0.01). (b) PFD group. The stable/improved group tended to have longer survival, but the difference was not significant (log-rank test; p = 0.06). (c) NTD group. The stable/improved group had a significantly better prognosis compared with the worsened group (log-rank test; p < 0.01).
In the case of NTD, the worsened group had a significantly higher incidence of AE than the stable/improved group (Gray test; p < 0.01), but there was no significant difference in the incidence of AE in PFD and the whole group (Figure 5).

Time to the first AE in the stable/improved group and the worsened group as classified by a 5% change in %FVC following antifibrotic therapy. (a, b) There was no significant difference in the incidence of AE between stable/improved and worsened groups in PFD and the whole group. (c) The worsened group had a higher incidence of acute exacerbations than the stable/improved group as classified by a 5% change in %FVC at 6 months following NTD therapy in patients with IPF (Gray-test; p < 0.01).
On the other hand, in terms of %FVC decline prior to the therapy and a slow rate of FVC decline, there was no significant difference in incidence of AE between stable and worsened groups and responder and non-responder groups, respectively (Figure S2, Figure S3).
Prognostic factors in IPF patients treated with antifibrotic therapy
We examined the prognostic factors in all populations using a Cox proportional hazards analysis (Table 2). In the univariate analysis, BMI, PaO2, FVC (L), and “stable/improved” status as defined by delta (Δ)FVCfollowing 6m, were significant factors. Subsequent to this analysis, ΔFVCfollowing 6m in the “stable/improved” group [hazard ratio (HR) 0.43; p = 0.02] had good prognosis after multivariate Cox proportional hazards analysis. In this study, %FVC decline prior to treatment (Δ%FVC
Risk factors for respiratory-related deaths following treatment with antifibrotic therapy.
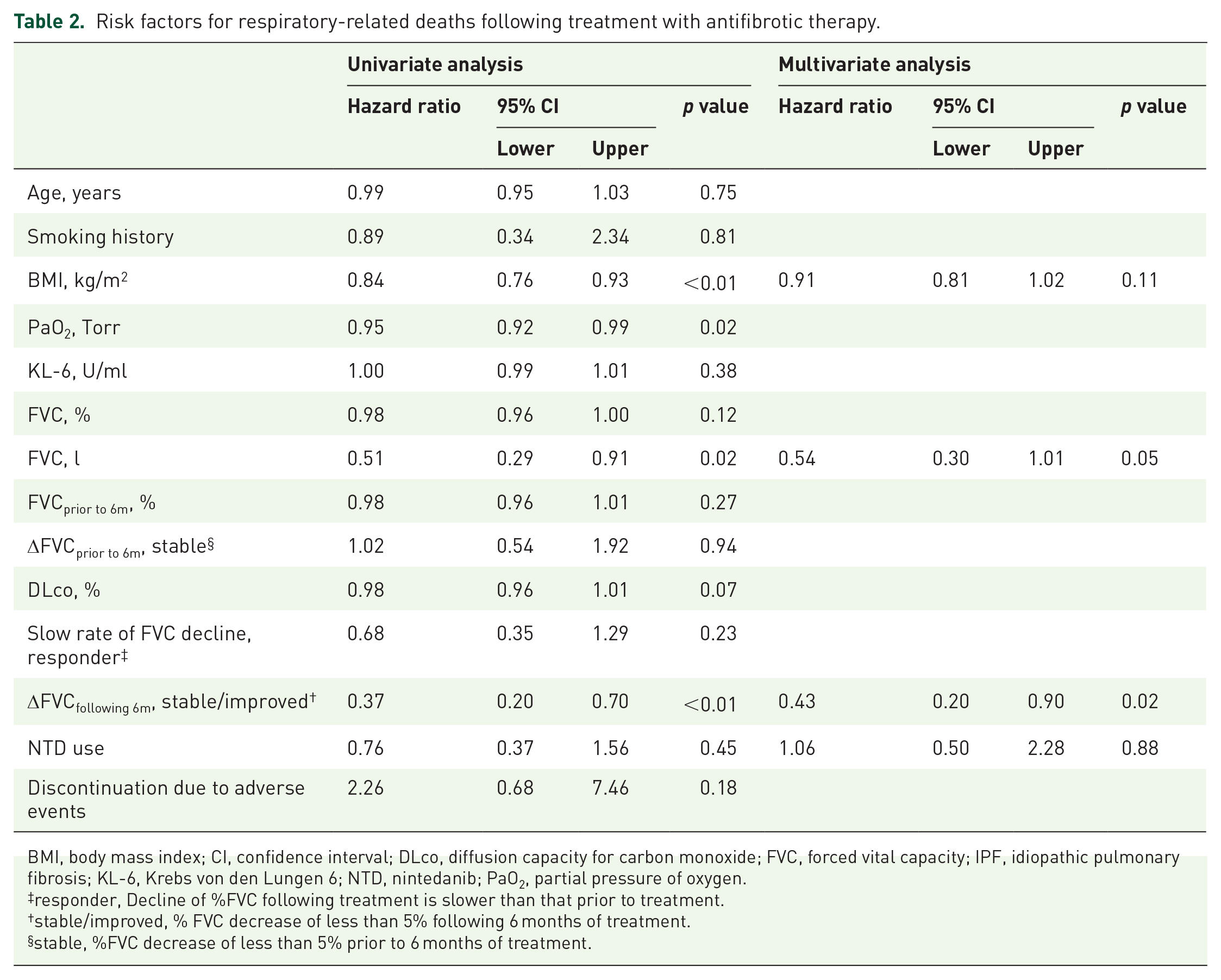
BMI, body mass index; CI, confidence interval; DLco, diffusion capacity for carbon monoxide; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis; KL-6, Krebs von den Lungen 6; NTD, nintedanib; PaO2, partial pressure of oxygen.
responder, Decline of %FVC following treatment is slower than that prior to treatment.
stable/improved, % FVC decrease of less than 5% following 6 months of treatment.
stable, %FVC decrease of less than 5% prior to 6 months of treatment.
Comparison of clinical features between the stable/improved group and the worsened group following antifibrotic therapy
The clinical features between patients in the stable/improved (n = 73) and worsened (n = 32) groups based on a 5% change or more in %FVC following antifibrotic therapy are shown in Table 3. There were no significant differences in baseline characteristics of age, sex, smoking status, laboratory data, and discontinuation of treatment. However, the stable/improved group showed a significantly higher BMI (23.4 versus 20.4 kg/m2; p < 0.01) than the worsened group.
Comparison of clinical features between the stable/improved group and the worsened group.

Categorical data are presented as numbers (percentages) or medians (range).
6m, 6 months; BMI, body mass index; DLco, diffusion capacity for carbon monoxide; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis; KL-6, Krebs von den Lungen 6; NTD, nintedanib; PaO2, partial pressure of oxygen; SP-D, surfactant protein-D.
Discussion
In this study, we investigated whether the FVC change prior to and following the treatment with antifibrotic drugs had impact on patient survival. %FVC decline following treatment with antifibrotic drugs, especially with NTD, is a strong prognostic factor in IPF. However, importantly, an FVC decline prior to treatment and a slow rate of decline in FVC are not associated with survival outcomes for both antifibrotic drugs.
Maher reported the reasons given by pulmonologists for not treating IPF in an international survey. Those results showed that, when it is a stable disease, many pulmonologists waited before initiating treatment in patients with IPF. 12 In fact, there are studies showing that a decline in FVC has been reliably associated with decreased survival.13–16 Meanwhile, Biondini et al. reported that patients with more rapidly progressive disease as defined by rate of pretreatment FVC decline, appeared to gain greater beneficial effects from PFD within 6–12 months of drug initiation compared with those with slower progression. 17 We found that %FVC decline prior to treatment and a slow rate of decline in %FVC is not associated with survival outcome. Furthermore, we also showed that a decline in %FVC following treatment is a strong significant prognostic factor. Nathan et al. suggested that FVC change following treatment may not predict future lung function. 18 However, consistent with our study, Richeldi suggested that an FVC change after 12 months of treatment with nintedanib may have good impact on survival outcome. 19 Collectively, there may be no need to watch and wait until the disease progresses. In other words, physicians should look at their patients’ early response to treatment, then switch to other drugs, take part in clinical trials, or proceed with lung transplantation without delay if needed.
Prediction of disease course or survival in IPF remains of interest for clinicians and patients. Baseline and longitudinal clinical, functional, biological, and radiologic findings have been studied extensively as prognostic predictors.20–23 In this study, we showed that an early disease progression with a %FVC decline despite antifibrotic therapy were significantly associated with a poor prognosis. These data suggest that an early physiological evaluation following antifibrotic therapy is important to predict outcomes in patients with IPF.
Meanwhile, a %FVC decline following treatment with NTD is strongly associated with patient survival rather than treatment with PFD.
There is no study of a direct comparison of survival outcome following treatment with either NTD or PFD. Rochwerg et al. reported an indirect comparison of the two, which showed no significant difference in mortality between NTD and PFD using a network meta-analysis for treatment of IPF. 24 Fleetwood K et al. reported that NTD and PFD are effective at reducing lung-function decline, and that PFD may reduce the odds of experiencing a decline in percent predicted FVC by >/=10% compared with placebo in the first year of treatment. The results of their analysis also suggest that PFD improves survival. 25 Meanwhile, Loveman et al. also reported that the two treatments show beneficial effects, but, when compared indirectly, NTD appears to have a superior benefit on forced vital capacity. 26 Recently, two studies were published using statistical methods widely used for life data analysis. Fischer et al. included patients enrolled in the ASCEND and CAPACITY trials who met inclusion criteria and used analysis by Weibull distribution. 27 The latter study showed that patients with IPF have improved life expectancy if treated with PFD compared with treatment with supportive care. Similarly, Lancaster et al. analyzed pooled data from six trials of NTD. 28 Exploratory analyses based on extrapolation of survival data suggest that NTD also extends life expectancy in patients with IPF. Although the population of these two studies were different, the shape of the curve is similar to our study’s results in terms of the stable/improved group following treatment with either drugs. This indicates that NTD may have more impact on survival compared with PFD if the disease has stabilized/improved following treatment. On the other hand, patients who had significant FVC declines following antifibrotic treatment had bad prognosis, but it is unknown as to which factors influenced these results. BMI was reported to be a prognostic factor in several studies.29–31 Consistent with these data, our study shows that patients in the stable/improved group following treatment had higher BMI values, suggesting that BMI might be associated with poor survival outcome even after antifibrotic treatment. Further studies are needed to clarify these issues.
The incidence of AE might be associated with the previously mentioned results. AE is the leading cause of death in IPF patients, 32 and there are studies suggesting that NTD may reduce the risk of AE.33,34 These data suggest that reducing AE might lead to increased survival as well as maintenance of lung function. Moreover, Kondoh et al. reported that a rapid %VC decline following treatment with antifibrotic drugs is a risk factor for AE-IPF. 35 Consistent with this, the risk of a first AE was higher in the worsened group than in the stable/improved group in the NTD-treated patients in our study.
Our study has several limitations. First, it was a retrospective and relatively small study, although it was a multicenter one. Therefore, there were selection biases, such as a high degree of pulmonary function impairment, and comorbidities. Second, information was derived from a review of electronic medical records, and thus depended on the subjects to actively report adverse reactions to their healthcare providers. As a result, the true incidence of adverse reactions may have been underestimated in our cohort.
In conclusion, the %FVC decline following antifibrotic therapy is important for survival outcome in our real-world cohort study. Concerning the disease based on FVC behavior prior to and following antifibrotic treatment, early treatment initiation may be beneficial for IPF.
Supplemental Material
Aono_et_al_Supplementary_Figures – Supplemental material for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis
Supplemental material, Aono_et_al_Supplementary_Figures for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis by Yuya Aono, Yutaro Nakamura, Masato Kono, Hidenori Nakamura, Koshi Yokomura, Shiro Imokawa, Mikio Toyoshima, Hideki Yasui, Hironao Hozumi, Masato Karayama, Yuzo Suzuki, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, Naoki Inui and Takafumi Suda in Therapeutic Advances in Respiratory Disease
Supplemental Material
Author_Response_1 – Supplemental material for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis
Supplemental material, Author_Response_1 for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis by Yuya Aono, Yutaro Nakamura, Masato Kono, Hidenori Nakamura, Koshi Yokomura, Shiro Imokawa, Mikio Toyoshima, Hideki Yasui, Hironao Hozumi, Masato Karayama, Yuzo Suzuki, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, Naoki Inui and Takafumi Suda in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_1_v.1 – Supplemental material for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis
Supplemental material, Reviewer_1_v.1 for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis by Yuya Aono, Yutaro Nakamura, Masato Kono, Hidenori Nakamura, Koshi Yokomura, Shiro Imokawa, Mikio Toyoshima, Hideki Yasui, Hironao Hozumi, Masato Karayama, Yuzo Suzuki, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, Naoki Inui and Takafumi Suda in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_2_v.1 – Supplemental material for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis
Supplemental material, Reviewer_2_v.1 for Prognostic significance of forced vital capacity decline prior to and following antifibrotic therapy in idiopathic pulmonary fibrosis by Yuya Aono, Yutaro Nakamura, Masato Kono, Hidenori Nakamura, Koshi Yokomura, Shiro Imokawa, Mikio Toyoshima, Hideki Yasui, Hironao Hozumi, Masato Karayama, Yuzo Suzuki, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, Naoki Inui and Takafumi Suda in Therapeutic Advances in Respiratory Disease
Footnotes
Author contribution(s)
Conflict of interest statement
Y. Nakamura and T. Suda received an honorarium and research funding from Boehringer Ingelheim Co., Ltd. All other authors declare no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
The reviews of this paper are available via the supplemental material section.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.