
Abstract
Pirfenidone is the first antifibrotic agent to be approved for the treatment of pulmonary fibrosis. Idiopathic pulmonary fibrosis (IPF) is one of the idiopathic interstitial pneumonias with the worst prognoses; approximately half of patients die within 3–5 years, and the need for an effective treatment has been unmet until recently. The etiology of IPF is still unknown and its pathogenesis is poorly understood. Anti-inflammatory drugs, such as corticosteroids and some immunosuppressants, have been empirically used to treat IPF, although they have not been objectively proven to be effective by large-scale randomized, controlled trials. Pirfenidone is an agent that can inhibit the decline of forced vital capacity (FVC)/vital capacity (VC) and that thereby can be hoped to decrease the mortality rate. The number of clinical trials of pirfenidone completed, ongoing, or planned is growing, and the present status of pirfenidone as treatment for IPF is summarized in this review.
Keywords
Introduction
Pirfenidone is the first antifibrotic agent approved for treatment of pulmonary fibrosis. Idiopathic pulmonary fibrosis (IPF) is one of the idiopathic interstitial pneumonias with the worst prognoses. Approximately half of patients die within 3–5 years. The need for an effective treatment has been unmet until recently [Azuma, 2010]. The etiology of IPF is still unknown and its pathogenesis is poorly understood. Anti-inflammatory drugs, such as corticosteroids and some immunosuppressants, have been empirically used to treat IPF, although they have not been objectively proven to be effective by large-scale randomized, controlled trials [Spagnolo et al. 2010]. IPF is a disease entity characterized by reticular opacity predominantly in the subpleural area and cystic lesions identified by high-resolution computed tomography (HRCT) scan and by the destruction of alveolar structure accompanied by collagen deposition identified by surgical lung biopsy. Although honeycombing in the subpleural area is a typical feature for IPF/usual interstitial pneumonia (UIP), various other factors affect its prognosis [Raghu et al. 2011]. Based on the current clinical understanding of IPF and the core concepts of its pathogenesis, the antifibrotic agent pirfenidone is one of the agents that can inhibit the decline of forced vital capacity (FVC)/vital capacity (VC) and that thereby can be hoped to decrease the mortality rate [du Bois et al. 2011]. The number of clinical trials of pirfenidone completed, ongoing, or planned is growing. The present status of pirfenidone as treatment for IPF is summarized in this review.
Clinical trials for IPF
Pilot studies
The benefit of pirfenidone treatment for pulmonary fibrosis was first observed in a patient whose pulmonary fibrosis developed as a result of bleomycin treatment for bladder cancer. An open-label 2-year study in 54 IPF patients was performed by Raghu and colleagues [Raghu et al. 1999], and an open-label 1-year study was performed in Japan [Nagai et al. 2002]. These reports showed that pirfenidone stabilized pulmonary function. In Mexico, a small-scale study in patients with Hermansky–Pudlak syndrome was also reported [Gahl et al. 2002]. Measurement of pulmonary function parameters, i.e. FVC, forced expiratory volume in one second (FEV1), total lung capacity (TLC), and carbon monoxide diffusion capacity (DLCO) showed a slowing of deterioration in patients with a baseline %FVC >50%.
Japanese trials
The first double-blind, randomized, controlled trial of IPF was a phase II trial conducted in Japan (2000–2002). At that time, there was no consensus on the most appropriate endpoint to use for the evaluation of therapeutic effect of IPF. After trial and error, the change in the lowest saturation of peripheral oxygen (SpO2) by pulse oximetry during a 6-min steady-state exercise test (6MET) at constant speed using a treadmill was selected as the primary endpoint. This endpoint was suitable for evaluation of drug efficacy over a relatively short period (1-year) [Azuma et al. 2005]. Eligible patients were diagnosed with ‘chronically progressive idiopathic interstitial pneumonia’ as defined by the third edition of the diagnostic criteria for IIPs in Japan at that time [Homma et al. 1992] and had arterial partial pressure of oxygen (PaO2) at rest of ≥70 Torr and SpO2 during exercise ≤ 90%. Although this trial was originally set up for a 1-year period, interim analysis at 6 months showed that all five cases with symptoms of acute exacerbations were in the placebo groups. The trial was terminated at 9 months and key opened according to the recommendation of the Data and Safety Monitoring Board (DSMB). As a result, no significant difference was observed between the two groups in the primary endpoint (change in the lowest SpO2) in the full analysis set (FAS), but a significant improvement was noted in the pirfenidone group compared with the placebo group at both 6 and 9 months in those patients who were able to complete the walking test during the trial. Furthermore, a significant change in VC indicating efficacy was observed at 9 months. However, serum marker levels, PaO2, and DLCO did not change significantly. Major side effects were photosensitivity and anorexia [Azuma et al. 2005].
A phase III trial was then conducted to determine effective dosages and reproducibility of the results of the phase II trial (2004–2006). The diagnosis of IPF was in accordance with the ATS/ERS Consensus statement [American Thoracic Society, 2000] and the fourth edition of the clinical diagnostic criteria for IIPs in Japan [Japanese Respiratory Society, 2004]. Eligible patients were adults (20–75 years of age) meeting the following SpO2 criteria: oxygen desaturation of ≥5% difference between resting SpO2 and the lowest SpO2 during a 6MET, and lowest SpO2 during the 6MET of ≥85% while breathing air. Eligible patients were assigned to three groups: high-dose (1800 mg/day), low-dose (1200 mg/day), and placebo groups, at the ratio of 2:1:2 (108, 55, and 104 patients, respectively) for the FAS. The 52-week observation period included an initial 4 weeks for dose titration. The primary endpoint was initially the lowest SpO2 during the 6MET but later became the change in VC under blinding according to the recommendation of the DSMB. The secondary endpoints were progression-free survival (PFS) time (with disease progression defined as more than 10% decrease in VC and/or death) and change in the lowest SpO2 during the 6MET. Significant differences were observed in the change in VC between the pirfenidone-treated groups and the placebo group in the FAS population [Taniguchi et al. 2010]. Among the secondary endpoints, PFS time was significantly prolonged in the high-dose group, providing the first evidence that a single drug improves PFS time in patients with IPF. However, there was no significant between-group difference in the lowest SpO2 using the FAS in this study. Major side effects were photosensitivity, anorexia, and elevation of γ-GTP [Taniguchi et al. 2010].
These phase II and III trials were conducted by members of the interstitial lung diseases committee of the Japanese Ministry of Health, Labour and Welfare and were sponsored by Shionogi & Co., Ltd. In October 2008, pirfenidone was approved for use against IPF for the first time in the world. A post hoc analysis of the data from the phase III study demonstrated a favorable response to pirfenidone treatment (i.e. a change in VC) in the subgroup characterized by ≥70% of predicted VC and <90% of SpO2 during the 6MET. We named this pirfenidone-responsive group the ‘phenostage’ as a candidate term for the subpopulation, which means not ‘responsible phenotype’ but ‘responsible period’ for therapeutic intervention [Azuma et al. 2011]. Subjective parameters, such as cough and dyspnea, as measured by the Fletcher, Hugh-Jones (F, H-J) classification [Fletcher, 1952] were also significantly improved in the treatment group (Figure 1). Thus, use of pirfenidone was apparently more efficacious in the early stage than in the advanced stage of IPF [Azuma et al. 2011].

Temporal changes in Fletcher, Hugh-Jones (F, H-J) classification in subpopulations of phase III trial.
The CAPACITY trials and the ASCEND study
Twin large-scale, well-controlled phase III trials named CAPACITY1 (Study 006) and CAPACITY2 (Study 004) were mainly conducted in North America and European countries (by InterMune, Inc.), respectively, beginning in 2006 [Noble et al. 2011]. Using change in %FVC at 72 weeks compared with baseline values as the primary endpoint, the study showed a statistically significant difference between the placebo group and treatment (2403 mg per day) group in Study 006, but not in Study 004 (Table 1). The European Medicines Agency (EMA) approved the use of pirfenidone for IPF in March 2011 based on the results of Study 004 and the Japanese phase III trial. However, the Food and Drug Administration (FDA) did not approve pirfenidone, because Study 006 failed to meet its primary endpoint. The post hoc analysis showed that the rates of decline in %FVC at week 72 in the placebo-treated group of Study 006 had been lower than those of other groups in Study 006 or a large trial of interferon-γ 1b [King et al. 2009], further strengthening the hypothesis of an attenuated FVC decline in the placebo group in Study 006. Moreover, assessment of baseline characteristics in Study 004 and Study 006 revealed that the placebo group in Study 006 had a greater proportion of patients with obstructive airway disease, which characteristically is associated with reduced FVC decline. These baseline inconsistencies leading to variability in rates of FVC decline in patients with IPF could partly account for the attenuated rate of FVC decline in the placebo group in Study 006 [Noble et al. 2011]. Acknowledging this shortcoming, an additional trial, ASCEND, was planned and excluded all patients with chronic obstructive pulmonary disease [ClinicalTrials.gov identifier: NCT01366209].
Comparison of mean change from baseline in %FVC in 004 and 006 of the CAPACITY trials

A Cochrane Systematic Review showed that only pirfenidone had efficacy for IPF when PFS was the endpoint (Figure 2), as in the trials in Japan and part of the CAPACITY trials [Spagnolo et al. 2010]. Decline in VC in the two trials in Japan was mild in the pirfenidone groups in contrast with the placebo groups (n = 314, mean difference 0.08 l, 95% CI 0.03–0.13, p = 0.00065) [Spagnolo et al. 2010].

Progression-free survival in three trials (n = 1046) was significantly different between pirfenidone and placebo groups (hazard ratio 0.7, 95% confidence interval 0.56–0.88, p = 0.002) (Reproduced with permission from Spagnolo et al. [2010])
Mechanisms of antifibrosis
Pirfenidone (5-methyl-1-phenyl-1H-pyridin-2-one) was discovered by Solomon B. Margolin of Marnac, Inc. (TX, USA) [Margolin and Lefkowitz, 1994]. Although originally shown to be an antifibrotic, pirfenidone has other activities including anti-inflammatory and antihydroxy radical activities [Azuma, 2010]. However, a high concentration of pirfenidone is needed to demonstrate efficacy in vitro, and there are no available in vitro assays for this drug. On the other hand, there are in vivo assays for pirfenidone. These in vivo assays not only demonstrate the antifibrotic activity of pirfenidone in the bleomycin-induced pulmonary fibrosis model, but also in the liver fibrosis, renal sclerosis, and sclerosing dermatosis models [Schaefer et al. 2011]. The antifibrotic activity of pirfenidone demonstrated in various organ fibrosis models is mirrored by the demonstration of pirfenidone inhibition of transforming growth factor beta (TGF-β) production. Thus, the inhibition of TGF-β production is thought to be crucial to antifibrotic activity. In addition, pirfenidone prevents the death of septic mice; this anti-inflammatory activity of pirfenidone might be due to the amelioration of the effect of tumor necrosis factor alpha (TNF-α) [Oku et al. 2002]. Oku and colleagues revealed that prednisolone did not inhibit the fibrotic process, but that on the other hand, pirfenidone clearly inhibited both fibrosis and inflammation in the bleomycin-induced mice model. In addressing the question of how different mechanisms contribute to fibrotic process of these mice, they found that pirfenidone (unlike prednisolone) inhibited the elevation of profibrotic cytokines, such as TGF-β and basic fibroblast growth factor (bFGF), and prevented deterioration of interferon gamma (IFN-γ) to maintain Th1/Th2 balance (Figure 3) [Oku et al. 2008; Oku, 2009]. There are at least two considerations from these results. First, attenuation of inflammation does not always contribute to subsequent antifibrotic activity. Second, the antifibrotic activity might be due to a unique antifibrotic mechanism of pirfenidone which might be independent of the anti-inflammatory mechanism. We continue to investigate the mechanism of pirfenidone’s effect on the fibrotic process in these models.
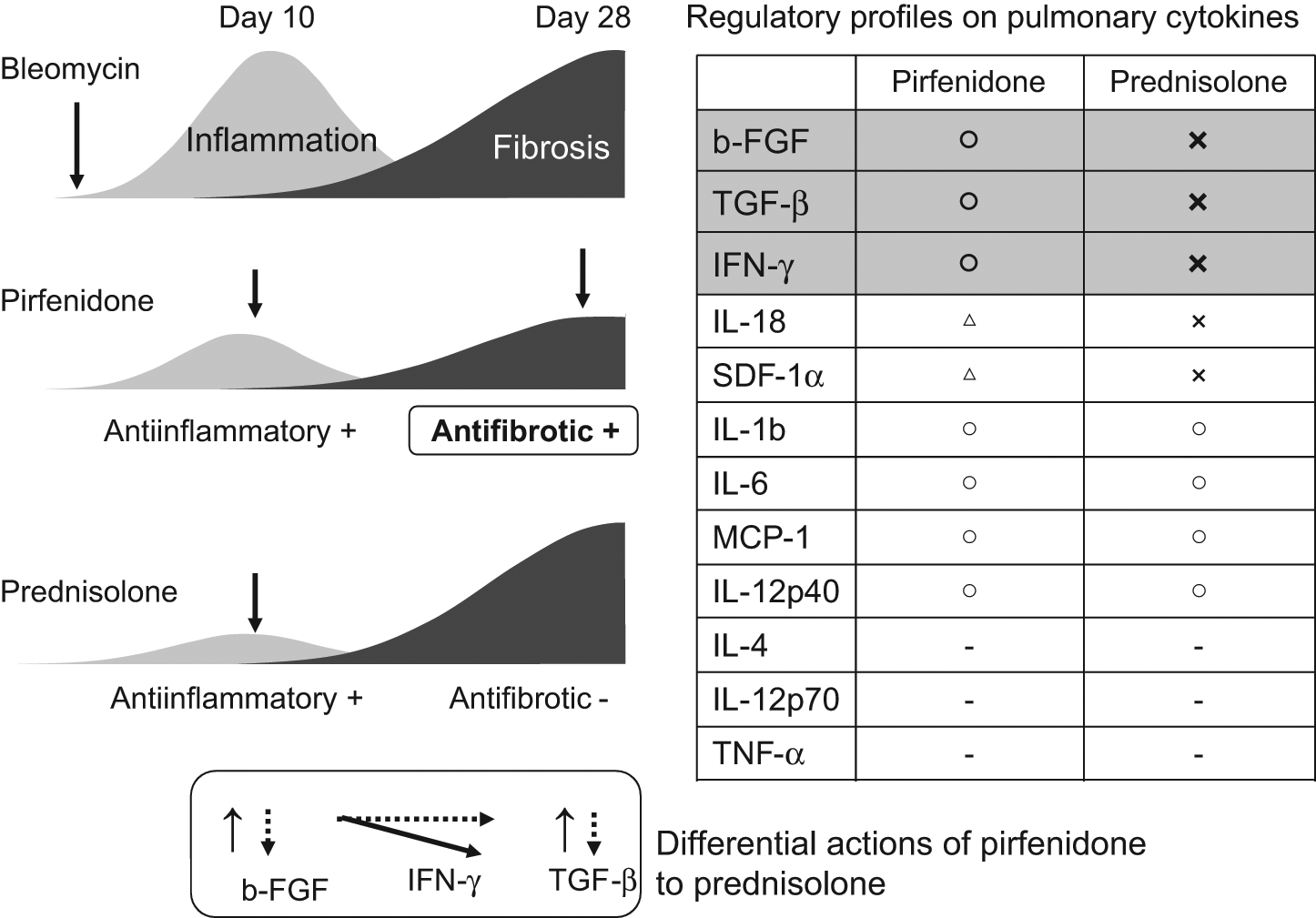
Differential regulatory profiles of pirfenidone and prednisolone in bleomycin-induced murine pulmonary fibrosis.
In addition, pirfenidone does not normally inhibit either humoral or cellular immunity which be expected to rarely induce an immunosuppressive condition by its use [Shionogi & Co. Ltd, 2008]. This characteristic can be an advantage of the treatment for IPF in clinical practice.
How pirfenidone can be used for IPF treatment: the utility of the new evidenced-based guidelines
Pirfenidone (Pirespa®) was launched in Japan in December 2008 and in the European Union (EU) (Esbriet®) in the autumn of 2011. Under what circumstances should pirfenidone be used clinically as an antifibrotic agent?
Despite the approvals, the recent publication of evidenced-based (EB) guidelines that were established by the American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and Latin American Thoracic Association (ALAT) concluded that there is no definitive treatment for IPF at present [Raghu et al. 2011]. The guidelines committee set up a grading system, mainly for common diseases, known as the Grading of Recommendations Assessment Development and Evaluation (GRADE) system, to evaluate the quality of evidence taking into account factors such as the number of patients. No consideration has been given to the adequacy of the GRADE system for the evaluation of evidence of the efficacy of new rare lung disease treatments. Further evaluation of the adequacy of the GRADE system for the assessment of IPF treatment is needed. In general, this author’s view is that ‘no evidence’ neither indicates proof of efficacy nor proof of inefficacy. EB guidelines are useful for the management of common diseases, not rare diseases. From this viewpoint, neither clinical practitioners nor patients with IPF will find the new EB guidelines useful. Pirfenidone treatment has been assigned a ‘weak against’ recommendation according to the new EB guideline. However, this author believes that pirfenidone should be used to search for the responsive subpopulation of patients with IPF among the larger population with heterogeneously and progressively pulmonary fibrosis. Further investigation is needed to discover new criteria for selecting patients who might benefit from IPF treatment.
Is IPF a single disease entity? The author believes that IPF is identifiable by radiological and pathological findings, particularly by findings of honeycomb lung. The new EB guideline describes the importance of radiological features in the diagnosis of IPF. Generally its 50% survival is known to be 3–5 years; however, IPF might have multifactorial etiologies, each of which might affect its differential progression and differential responses to the treatment. In the clinical trials, we found that there were both good and poor responders to pirfenidone. We, medical doctors, are obligated to treat each IPF patient individually using effective strategies based on the results of well-designed clinical trials. So subset analysis of good responders among IPF patients will continue. IPF patients with mildly or moderately impaired FVC/VC are better responders to pirfenidone treatment for 3–12 months [Azuma et al. 2011]. In other words, every IPF patient can possibly benefit from pirfenidone when their %VC is more than 70% and oxygen desaturation is under 90% during the six-minute walking test (6MWT), that is, when the disease is in what has been defined by the present author as the ‘phenostage’ indicating an early stage of IPF characterized by less impairment of static and dynamic pulmonary functions instead of phenotype prescribed by genotyping. In this stage, cough symptoms and dyspnea are also improved by pirfenidone [Azuma et al. 2011] (Figure 1). Evaluation of responsiveness to pirfenidone should improve the efficacy of pirfenidone in IPF treatment. A 5% change of VC in 3 months is a predictor of 1-year efficacy [Taniguchi et al. 2011]. Prognostic factors of IPF can be established by including a placebo group of IPF registered in previous clinical trials [du Bois et al. 2011]. Statistical analysis revealed that (1) age at entry, (2) previous history of hospitalization due to respiratory diseases, (3) FVC, and (4) change in FVC in a steady-state period were prognostic indicators of upcoming mortality within a year. Indicator (4) but not (1)–(3) can be improved by treatment intervention. Pirfenidone is the only agent proven to inhibit the decline in FVC/VC in independent clinical trials, and so we can target FVC decline with pirfenidone to reduce mortality rates. Furthermore, we will be looking for agents that can be combined with pirfenidone to reduce the decline in FVC. Combined therapy with pirfenidone and upcoming other agents will be important. The ultimate goal is to halt the decline in pulmonary function resulting in destruction and fibrosis. Since the pathogenesis of IPF might be heterogeneous, the final target of IPF treatment is multiple pathways that lead to fibrosis progression. This concept is similar to that of cancer treatment.
Implementation of clinical trials for IPF (even IPF with the same features as UIP) has been difficult due to its relatively low prevalence and heterogeneous progression. In this regard, the appropriate primary efficacy endpoint has not been established. Because the significance of the primary endpoint was reached and sometimes lost even in trials by the same protocol, feature of patients is valuable due to various prognostic factors, as not only change of static pulmonary functions, but also the complications of acute exacerbation, lung cancer and pulmonary hypertension, as well. IPF is basically characterized by restrictive lung impairment, defined as decreased VC or FVC and leads to irreversibly progressive decline in gas exchange, such as the decline in DLCO. In this regard, selection of the primary endpoint is an important and controversial issue. Since mortality rate is an essential parameter for predicting treatment efficacy, increasing the number of patients and the time period of observation will be required to get significant results. At present, change in the FVC/VC ratio is an alternative surrogate marker of survival in patients with IPF [Taniguchi et al. 2011]. Change of FVC has been considered predictive of outcome. Categorical change of 5–10% in FVC in 6 months has been found to be a potential predictor of mortality in the following year, based on both clinical trials and IPF cohorts data [Richeldi and du Bois, 2011; Zappala et al. 2010]. So we currently believe that change in FVC/VC is the most reliable predictor of survival.
Future aspects
Introduction of pirfenidone to the field of IPF treatment was a small step forward and it has led us to new insights. We are collecting clinical experience, efficacy, and safety data in Japanese IPF patients and some patients from other countries. In 3 years, we have collected data on the treatment of progressive constrictive lung fibrosis with systemic sclerosis, rheumatoid arthritis, and chronic hypersensitivity pneumonia. This experience has led us to wonder what we need to do to get regulatory approval for a clinical trial in patients with these rare lung diseases. The disease entity ‘chronic progressive lung fibrosis of any origin’ might be generally acceptable for clinical trials of antifibrotic agents, in which changes in pulmonary function and gas exchange affecting the survival benefit of treatment are to be evaluated [du Bois et al. 2011]. This author realizes the necessity to evaluate the survival benefit of (1) pirfenidone alone in advanced IPF and (2) combined with other promising agents such as N-acetylcysteine (NAC) in patients with IPF. The PANTHER study failed to show any significant efficacy of NAC combined with prednisolone and azathioprine [McGrath and Millar, 2011]. The DSMB recommended discontinuation of NAC combination therapy, but not of NAC monotherapy. Nevertheless, the efficacy of NAC monotherapy remains to be proven [Homma et al. 2011]. Before promoting the use of pirfenidone combined with other agents, we need to examine carefully factors affecting the efficacy of each treatment. We will be looking not only for improvement in FVC/VC but also for effects on pulmonary arterial hypertension, carcinogenesis, neovascularization, thromboembolism, and acute exacerbations of IPF.
Only combination therapies may maximize survival benefit in the future, as is the case in cancer treatment. This author recognizes that the conflict between the need for a large sample (in order to obtain approval for a clinical trial) and the heterogeneity of rare lung diseases poses a dilemma.
Summary and key messages
Three years have passed since the introduction of pirfenidone as treatment for IPF. Over 2500 patients with IPF have been treated with pirfenidone in Japan. Adverse events, such as photosensitivity and gastrointestinal discomfort, were made tolerable by the use of countermeasures in the majority of cases. It is accepted that analysis of data from some clinical trials identified an IPF-responsive subpopulation (‘phenostage’). It is also accepted by this author that pulmonary function is not always usable as a primary endpoint if combined with obstructive functional impairment [Noble et al. 2011; Cottin et al. 2005]. Although IPF is a complicated disease, it is clear that pirfenidone is the first promising agent for IPF treatment; the author is encouraged that this agent has further value in the field of IPF treatment.
Footnotes
This research is partly supported by the Diffuse Lung Diseases Research Grant from the Ministry of Health, Labour and Welfare, Japan.
Arata Azuma has received consulting fees/honoraria from Shionogi & Co. Ltd, Boehringer Ingelheim, Bayer, Novartis Pharma, and Pfizer and speaker’s fees from Shionogi & Co. Ltd and Eisai Co. Assistance with writing in English was received during the preparation of this manuscript.