
Abstract
Sarcoidosis is a systemic inflammatory disease of unknown etiology that is characterized by the presence of granulomatous inflammation in affected tissues. It can affect essentially any organ system but shows a predilection for the lungs, eyes, and skin. Accurate epidemiological data are not available in the USA, but sarcoidosis is considered a ‘rare disease’ (prevalence less than 200,000). However, recent epidemiologic studies indicate that regional prevalence is much higher than previously estimated, especially among African American women. Additionally, mortality rates of patients with sarcoidosis are increasing by 3% per year over the past two decades. The most common causes of death are attributed to progressive lung disease and cardiac sarcoidosis, and the health of the patients is further compromised by other systemic manifestations. As such, the management of sarcoidosis requires a collaborative multidisciplinary approach. We aim to discuss the principles of managing sarcoidosis, including standards of care relating to pulmonary disease as well as recent advances relating to the detection and treatment of extrapulmonary manifestations.
Keywords
Introduction
Sarcoidosis is a systemic inflammatory disease of unknown etiology that is characterized by the presence of granulomatous inflammation in affected tissues. It can affect essentially any organ system but shows a predilection for the lungs, eyes, and skin. Accurate epidemiological data are not available in the USA, but sarcoidosis is considered a ‘rare disease’ (prevalence less than 200,000). However, recent studies indicate that regional prevalence is much higher than previously estimated, especially among African American women [Tukey et al. 2013]. Additionally, mortality rates of patients with sarcoidosis have increased by 3% per year over the past two decades [Swigris et al. 2011]. The most common causes of death are attributed to progressive lung disease and cardiac sarcoidosis (CS) [Swigris et al. 2011]. The health of these patients is further compromised by other systemic manifestations. As such, the management of sarcoidosis requires a collaborative multidisciplinary approach. We aim to discuss the principles of managing sarcoidosis, including standards of care relating to pulmonary disease as well as recent advances relating to the detection and treatment of extrapulmonary manifestations.
Pathogenesis
The immunopathogenesis of sarcoidosis is complex and not completely understood, and a full discussion of this topic is beyond the scope of this review. In brief, it is postulated that exposure to an unknown environmental antigen in a predisposed individual leads to an abnormal type I immune response. Antigen presentation via major histocompatibility complex class II molecules leads to the activation and oligoclonal expansion of CD4+ T cells (Figure 1). These CD4+ T cells differentiate predominately into type 1 helper T cells (Th1 cells), which subsequently secrete interleukin-2 and interferon γ. This augments tumor necrosis factor α (TNFα) production by macrophages and potentiates the local immune response [Iannuzzi and Fontana, 2011]. TNFα signaling and cell-mediated immunity lead to the formation and sustainment of granulomas, the pathologic hallmark of sarcoidosis. Although granulomas may resolve with little consequence in most patients, pulmonary fibrosis develops in 20–25% of patients [Iannuzzi and Fontana, 2011]. The pathophysiology of fibrosis in pulmonary sarcoidosis remains poorly understood but likely involves a shift from Th1 to a type 2 T-helper (Th2) cell response [Patterson et al. 2012; Wynn, 2004].

In the patient who is genetically predisposed, antigen presentation leads to CD4 activation and proliferation. The type 1 T helper (Th1) immune response results in local oligoclonal proliferation of T cells, tumor necrosis factor α (TNFα) and other cytokine release, and granuloma formation. The final stage of the inflammatory response may take the form of disease resolution or fibrosis, and likely depends on antigen clearance, a process that may be impaired in those with sarcoidosis. APC, antigen-presenting cell; IFN, interferon; IL, interleukin; TGFβ, transforming growth factor β.
A single causative antigen has not been confirmed and it remains likely that exposure to a variety of different antigens could promote the disease in a susceptible person. Numerous associates have been identified in epidemiologic studies [Bresnitz and Strom, 1983; Eishi et al. 2002; Izbicki et al. 2007; Newman et al. 2004; Song et al. 2005]. Considering the ubiquitous nature of many of the incriminated antigens, it seems likely that variables affecting the host immune response may primarily drive disease pathogenesis. There is strong evidence to support the notion that predisposition to sarcoidosis and variability in disease phenotype are influenced by genetic factors [Crouser et al. 2012; Fischer et al. 2010; Iannuzzi et al. 2005; Rossman et al. 2003; Rybicki et al. 2005].
Detection of disease activity
Once the diagnosis of sarcoidosis is established, it is important to determine which organs are involved and which symptoms are attributable to the disease. As described in the following section, disease phenotype strongly influences decisions to treat. Therefore, it is important to confirm that the patient’s disease manifestations are indeed related to sarcoidosis and not to other causes.
A thorough history and physical exam is the first step toward identifying organ systems affected in patients with sarcoidosis. Many patients will complain of generalized symptoms, including fatigue, fever, and weight loss [ATS/ERS/WASOG, 1999]. Common pulmonary symptoms include dyspnea, cough, and chest tightness. The pulmonary exam is often normal even in those with active disease. A minority present with wheezing or crackles, the latter reflecting end-stage fibrosis. Skin exam can reveal a variety of changes, including erythema nodosum, plaques, lupus pernio (disfiguring induration and discoloration of the face), maculopapular rash, scar lesions and subcutaneous nodules [Lodha et al. 2009; Yanardag et al. 2003]. A thorough eye exam may show anterior or posterior uveitis, lacrimal gland swelling or optic neuritis [Baughman et al. 2010]. In all cases, a comprehensive review of systems and complete physical exam are required to identify evidence of other extrapulmonary disease manifestations.
Radiographic, physiologic, and serological testing is useful for assessing organ involvement. The American Thoracic Society (ATS), the European Respiratory Society (ERS) and World Association of Sarcoidosis and Other Granulomatous diseases (WASOG) guidelines recommend a series of initial tests in all patients diagnosed with sarcoidosis [ATS/ERS/WASOG,1999]. These tests include a chest X-ray (CXR) and pulmonary function test (PFT) to evaluate for pulmonary involvement. Common CXR findings include lymphadenopathy and numerous parenchymal manifestations. The latter may include small nodules, most often in a peribronchiolar distribution that favors the mid and upper lobes. Other common radiographic manifestations include large nodules and ground glass infiltrates which preferentially involve the upper lobes [Akbar et al. 2008]. In some individuals, pulmonary sarcoidosis progresses to fibrosis or fibrocystic changes that are often accompanied by bronchiectasis [Lynch et al. 2007]. The Scadding staging system (Table 1) is one approach for categorizing pulmonary sarcoidosis based upon these common CXR manifestations [ATS/ERS/WASOG,1999; Scadding, 1961]. PFTs typically demonstrates restrictive lung disease but obstruction is not uncommon and may related to extrinsic compression (e.g. from mediastinal disease) or intrinsic airway involvement.
Modified Scadding chest radiographic stages.

The presented stages are modified from the original Scadding stages as per the American Thoracic Society (ATS), European Respiratory Society (ERS) and World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) guidelines [ATS/ERS/WASOG, 1999; Scadding, 1961].
Screening for extrapulmonary disease is also recommended, as per the ATS/ERS/WASOG guidelines [ATS/ERS/WASOG, 1999]. Blood chemistries, including blood urea nitrogen, creatinine, calcium, and liver function panel should be obtained to evaluate for renal dysfunction, hypercalcemia, and hepatic abnormalities. In those with altered renal function or with a history of nephrolithiasis, urine calcium is also recommended to evaluate for hypercalciuria. An ophthalmologic exam should be performed to evaluate for findings consistent with ocular involvement. Finally, an electrocardiogram (ECG) should be obtained at baseline to screen for evidence of possible cardiac involvement [ATS/ERS/WASAOG, 1999]. Additional cardiac testing, including 24 h Holter monitor, echocardiogram, and other imaging modalities, further enhances the detection of cardiac disease and will be discussed below [Mehta et al. 2008]. Further organ-specific testing should be directed by history and physical.
Organ assessment tools have been developed to assist in the clinical identification of individual organ involvement. The ACCESS (A Case Control Etiologic Study of Sarcoidosis) group developed an assessment tool that categorizes organ-specific abnormalities into definite, probable, or possible involvement based upon clinical assessment and widely available tests [Judson et al. 1999]. This tool was not intended to establish a diagnosis; rather it is used to predict the extent of organ involvement in those with biopsy-confirmed sarcoidosis. Since the tool was developed in 1999 there have been a number of new developments, including the evolution of positron emission tomography (PET) scans and magnetic resonance imaging (MRI) that improve the detection of disease. A revised version of the ACCESS organ assessment tool that will account for these and other innovations is under development by a team of experts affiliated with WASOG [Judson et al. 2014].
Indications for treatment
The decision to treat patients with sarcoidosis is often complex due to the lack of evidence-based indications for treatment and the potential for serious side effects of immunomodulating agents. Symptoms, disease course, and the threat of vital organ dysfunction should be carefully weighed against the risks attendant to therapy, and the clinician should be careful to confirm that the patient’s symptoms are indeed related to sarcoidosis. Fortunately, many patients diagnosed with sarcoidosis will experience spontaneous disease regression or stabilization and will not require treatment. Scadding originally described this finding in 136 patients that he followed over 5 years [Scadding, 1961]. He found that 97% of patients with stage 1 disease were asymptomatic without treatment 5 years from diagnosis while 58% of patients with stage 2 disease and 25% of patients with stage 3 disease were asymptomatic in the absence of treatment at 5 years [Scadding, 1961]. Data from the ACCESS cohort and from the Mana cohort in Spain confirmed that the majority of patients presenting with stage I disease do not progress, at least within 2 years [Baughman et al. 2001; Mana et al. 1994].
For this reason, a period of close observation is reasonable to determine the course of disease in an individual prior to considering therapy. However, some patients require immediate treatment, including those presenting with cardiac involvement, central nervous system disease, uveitis, severe hypercalcemia, disfiguring cutaneous manifestations (i.e. lupus pernio), upper respiratory obstruction, or severe pulmonary impairment [ATS/ERS/WASOG, 1999; Iannuzzi and Fontana, 2011; Lazar and Culver, 2010].
The threshold for initiating treatment for pulmonary sarcoidosis is not clearly defined. Most experts agree that steady progression of disease, as reflected by an objective loss of function over time or the development of life-altering symptoms, warrants a treatment trial. In this regard, a decline in the functional vital capacity (FVC) of 10% or diffusing capacity for carbon monoxide (DLCO) of 20% is considered significant [Lazar and Culver, 2010; Lynch et al. 2007]. In lieu of objective changes in lung function, it is reasonable to consider treating those who are symptomatic. However, other etiologies for the symptoms should be considered before attributing them to active sarcoidosis.
Other features of the initial clinical presentation are helpful prognostically and may guide treatment. African Americans, older patients, patients with Scadding stage 2–4 disease with active pulmonary symptoms, and those presenting with extrathoracic manifestations tend to have a more severe, progressive course and may benefit from therapy [ATS/ERS/WASOG, 1999; Lazar and Culver, 2010]. In contrast, patients with Löfgren’s syndrome (acute onset hilar lymphadenopathy, polyarthritis, and erythema nodosum) have a very favorable prognosis and often do not require treatment. Indications for the treatment of extrapulmonary disease are guided by the severity of illness and patient symptoms. Table 2, from a recent review by Iannuzzi and Fontana, provides a useful guide to treating various disease manifestations [Iannuzzi and Fontana, 2011].
General treatment recommendations for various sarcoidosis manifestations.

DLCO, diffusing capacity for carbon monoxide; FVC, functional vital capacity; TNFα, tumor necrosis factor α.
How to treat
First-line therapy
Glucocorticoids are the standard initial therapy for sarcoidosis. The typical starting dose is between 20 and 40 mg of prednisone per day [ATS/ERS/WASOG, 1999]. Response to treatment should be determined within the first 6 weeks and reevaluated regularly (i.e. every 3–6 months) thereafter to establish the minimum effective dose. The goal maintenance dose is the equivalent of prednisone 5–10 mg per day. Compared with daily administration, every other day dosing of the steroids is equally effective and may be advantageous in terms of allowing for more rapid tapering. The long-term goal is to eventually wean the steroids off.
Failure to respond to steroids or inability to wean prednisone to 10 mg or less within 3–6 months warrants a reassessment of the cause and consideration for adding a ‘steroid-sparing’ agent. Before doing so, patients should be evaluating for confounding issues. Such issues include noncompliance, irreversible fibrotic disease, the development of new complications (e.g. infection), or the possibility of occult pulmonary hypertension (PH).
Alternatives to glucocorticoids
Patients who cannot be weaned to prednisone dosages of less than 10 mg per day are appropriate candidates for steroid-sparing medications. Other potential candidates for these alternative agents include those with comorbidities that may be exacerbated by corticosteroids, such as diabetes mellitus, osteoporosis, difficult to control hypertension, or chronic nonhealing wounds. The appropriate drug regimen should be individualized based on the risk–benefit profile of the immunosuppressant and the degree of end organ involvement. Figure 2 shows a proposed algorithm for treatment of pulmonary sarcoidosis [Baughman et al. 2013]. This strategy has not been validated in a clinical trial. Few alternative immunosuppressants have been studied in randomized controlled trials. Additionally, none of these are approved by the US Food and Drug Administration for the treatment of sarcoidosis. Table 3 provides a summary of outcomes for clinical trials and level of evidence for commonly used immunomodulating drugs commonly used for the treatment of pulmonary sarcoidosis.

Proposed treatment strategy for treatment for pulmonary sarcoidosis. This figure is modified from a review by Baughman and colleagues [Baughman et al. 2013].
Outcomes of studies evaluating the use of alternative immunosuppressants in treatment of pulmonary sarcoidosis.

Level of evidence is based on grading recommendations published by the American College of Physicians. 1A = strong recommendation, high-quality evidence; 1B = strong recommendation, moderate-quality evidence; 1C = strong recommendation, low-quality evidence; 2A = weak recommendation, high-quality evidence; 2B = weak recommendation, moderate-quality evidence; 2C = weak recommendation, low-quality evidence.
No positive studies supporting its use in pulmonary sarcoidosis. There are multiple case reports of drug-induced sarcoidosis associated with use of etanercept for other indications. We strongly recommend against its use for treatment of sarcoidosis.
BAL, bronchoalveolar lavage; DLCO, diffusing capacity for carbon monoxide; FDG-PET, fludeoxyglucose positron emission tomography; FEV1, forced expiratory volume in 1 s; FVC, functional vital capacity; IL, interleukin; QOL, quality of life; TNFα, tumor necrosis factor α.
Local treatments
Whenever feasible, it is advantageous to employ local therapies to minimize the side effects of systemic treatments. For pulmonary, ocular, and cutaneous disease, local therapies may help to improve symptoms and reduce the cumulative dosage of systemic immunosuppression. High-dose inhaled corticosteroids may be beneficial in improving pulmonary function and cough. Inhaled budesonide at a dose of 800 μg twice daily resulted in improvement in lung function compared with placebo at 18 months in patients with symptomatic pulmonary sarcoidosis [Pietinalho et al. 1999]. In a 5-year follow-up study, those patients treated with inhaled budesonide required less frequent systemic corticosteroid use and had improved FVC and DLCO compared with the placebo group [Pietinalho et al. 2002]. Other trials evaluating inhaled fluticasone have shown improvements in cough, but did not demonstrate improved pulmonary function [Baughman et al. 2002a; du Bois et al. 1999].
Eye involvement occurs in around 25% of patients with sarcoidosis [Bradley et al. 2002]. Anterior uveitis is the most common manifestation and mild cases often respond to topical corticosteroids, often used in combination with mydriatic eye drops. More severe forms may require periocular corticosteroid injections. Failure to respond to local therapies mandates systemic immunosuppression. In this case, topical agents may continue to be used in addition to systemic therapy.
Cutaneous sarcoidosis occurs in up to a third of patients [Marchell and Judson, 2010]. Topical and intralesional steroids are first-line therapy for localized disease. Topical clobetasol proprionate 0.05% cream has been successful in treating papular skin disease [Volden, 1992]. Intralesional triamcinolone has also been shown to be effective in localized disease, and may be more effective than topical preparations [Haimovic et al. 2012]. Similar to ocular therapies, local therapy for cutaneous disease can also be used in conjunction with systemic agents and coadministration may facilitate tapering of the systemic corticosteroid dose [Haimovic et al. 2012]. Local therapy alone is not sufficient in patients with extensive cutaneous disease and treatment with systemic therapy is often warranted. When first-line and alternative therapies fail, TNFα antagonists have proven useful in this group of patients [Judson et al. 2008; Pariser et al. 2013; Stagaki et al. 2009]. Management in conjunction with a dermatologist is recommended while treating extensive or potentially disfiguring skin manifestations and facial lesions.
Acute pulmonary exacerbations of sarcoidosis
Acute pulmonary exacerbations of sarcoidosis (APES) was recently proposed to describe an acute worsening of pulmonary symptoms and a decline of over 10% in forced expiratory volume in 1 s (FEV1) or FVC from baseline [Panselinas and Judson, 2012]. The criteria for diagnosis of APES include duration of symptoms exceeding a month and the exclusion of alternative pulmonary diseases. Symptoms of APES are nonspecific but often include cough, wheezing, and dyspnea. Chest imaging is more useful to exclude other potential causes of pulmonary decline than to rule in an exacerbation.
The clinical utility of APES is designed to avert excessive long-term exposure to corticosteroids and other immunomodulating drugs. The recommended treatment of APES consists of a short course of corticosteroids or an increase in the current dose. In a study of 36 patients treated for APES, oral prednisone at 20 mg daily for a median duration of 21 days resulted in improvement in pulmonary symptoms and increase in FEV1 and FVC to the previous baseline [McKinzie et al. 2010]. We recommend prednisone 20–40 mg daily for 3 weeks with tapering of the dose thereafter. Patients should be reevaluated within 3–4 weeks of starting treatment to ensure appropriate weaning of the corticosteroid dose or initiation of a steroid-sparing agent if indicated. If APES recurs frequently, it would be reasonable to consider initiation of a steroid-sparing agent to attenuate recurrent disease flares.
Following disease course
Current guidelines recommend regular follow-up at 3–6-month intervals for patients with active disease, particularly following a change in the treatment regimen [ATS/ERS/WASOG, 1999]. Those with apparently inactive disease should be monitored at least yearly for 3 years. Patients with persistent radiographic changes should be followed indefinitely for recurrence of disease. However, routine monitoring of PFTs and chest imaging in inactive or ‘burnt out’ sarcoidosis is not necessary and should be reserved for those with new or worsening symptoms.
Routine surveillance for patients with active disease should include a thorough history and physical examination, chest imaging, and PFTs. The frequency of diagnostic testing should be guided by severity of disease and response to therapy. Further testing should be determined by the extent of extrapulmonary involvement and the presence of new symptoms. However, an ophthalmology evaluation is recommended annually regardless of whether there is eye involvement.
The gold standard metric for assessing disease progression and response to treatment is PFTs. FVC, FEV1, DLCO, and 6 min walk distance (6MWD) can be used to monitor progress. However, FVC is the most commonly used parameter in both clinical practice and research studies. A significant difference in FVC is considered an absolute change of 10%. However, the minimal clinically important difference in FVC has been shown to be 2–6% in some studies [du Bois et al. 2011]. Because such small changes in FVC can be within the range of test variation, we recommend changes in lung function always be correlated with clinical symptoms.
Other PFT variables such as FEV1, total lung capacity (TLC), DLCO, and 6MWD are less commonly used, but can provide useful information to the overall clinical picture. FEV1 can be helpful in monitoring those with obstructive physiology. DLCO can be useful for detecting the presence of PH or worsening gas exchange. However, it is not specific to sarcoidosis and can be affected by other variables. Therefore, changes in DLCO should be correlated with clinical findings. 6MWD can be helpful in appraising exercise tolerance and ambulatory oxygen requirements. It is most commonly used in the initial assessment of patients. It may also be beneficial in monitoring disease progress, but like DLCO can be influenced by nonpulmonary factors, including muscle strength, cardiac disease, anemia, and fatigue [Baughman and Lower, 2007]. Given these limitations and the lack of a superior metric of lung function, we routinely monitor full PFTs (i.e. spirometry, lung volumes, and DLCO) in patients with pulmonary disease. However, changes in lung function should be interpreted in conjunction with patient symptoms.
The chest radiograph is frequently used in the monitoring of pulmonary sarcoidosis progression. Its low cost and accessibility make it an appealing tool. Chest radiographs provide complementary information to PFTs, but they also detect changes in lung morphology (e.g. enlarging or cavitating nodules, bronchiectasis) that could have important clinical implications. Unfortunately, radiographic changes do not correlate well with changes in pulmonary function. Additionally, there is significant variability in interpretation of a given radiograph [Baughman et al. 2009b]. These factors limit the clinical utility of CXR for following disease progress. Nonetheless, they can provide useful information in the patient with new or worsening symptoms to evaluate for other causes, such as infection or pulmonary edema. The last iteration of the ATS/ERS/WASOG statement on sarcoidosis recommends annual CXRs in patients with active pulmonary sarcoidosis [ATS/ERS/WASOG, 1999].
Computed tomography (CT) is also used frequently in clinical practice and may be superior to CXR for following disease progression. The ability of CT scans to detect subtle parenchymal changes makes it a powerful tool to aid the detection and monitoring of disease activity. Unlike the chest radiograph, CT abnormalities have been shown to correlate with severity of PFT changes [Bergin et al. 1989; Drent et al. 2003; Erdal et al. 2012]. Additionally, severity of parenchymal changes on high-resolution CT has been shown to correlate with inflammatory activity of sarcoidosis [Oberstein et al. 1997]. However, assessment of disease severity by CT scans alone is not well standardized. Automated computerized image analysis offers a solution for this, but previous attempts to quantify severity of interstitial lung disease have been limited by only modest correlations [Best et al. 2003; Hartley et al. 1994; Rienmuller et al. 1991]. A newly developed quantitative computer-generated two-point correlation analysis offers better correlation and appears to more reliably assess pulmonary disease severity [Erdal et al. 2012]. This approach eliminates ‘background noise’ from normal lung structures (i.e. blood vessels, airways) to allow a more precise assessment of the parenchyma. Analysis generates a lung texture score, which strongly correlates with the inverse of FVC% predicted (r = −0.76), TLC (r = −0.61), and DLCO (r = −0.57) (p < 0.001 for all comparisons) [Kanis et al. 2004a]. Limitations of its routine use include the relatively higher cost of CT imaging (relative to CXR) and concerns about the long-term effects of radiation exposure. Further studies considering the utility of low-dose CT imaging in sarcoidosis are warranted. We recommend CT imaging be used selectively in patients with sarcoidosis.
Serum angiotensin-converting enzyme (SACE) levels have been classically associated with sarcoidosis. However, SACE levels are not always elevated in those with active sarcoidosis, and elevated SACE levels are not specific to sarcoidosis as this can be seen in other granulomatous diseases. Serial measurements of SACE levels have been suggested as a means for following clinical course in a subgroup of patients with sarcoidosis who present with elevated levels [Lieberman et al. 1983]. However, the lack of specificity and poor correlation with disease severity limits its usefulness as a clinical biomarker [Baughman et al. 1983].
Complex disease manifestations
End-stage lung disease
The clinical presentation and progression of pulmonary sarcoidosis is quite variable, ranging from spontaneous remission of disease to relentless progression to end-stage fibrocavitary disease. Approximately 5% of patients have radiographic evidence of fibrosis at diagnosis [Baughman et al. 2001]. Development of fibrosis and ongoing inflammation leads to bronchial distortion, cystic changes, and loss of normal lung architecture. These structural changes often result in significant symptoms and debility. They also place patients at risk of developing of serious complications, including bronchiectasis, PH, mycetoma formation, hemoptysis, and recurrent infections. Advanced lung disease accounts for most of the morbidity and mortality in sarcoidosis. In a retrospective study of 142 patients with sarcoidosis with radiographic stage IV disease, survival at 10 years was significantly worse than the general population [Nardi et al. 2011]. The cause of death was attributed to a respiratory-related complication in 75% of cases [Nardi et al. 2011].
Lung fibrosis in sarcoidosis is an irreversible process. The benefits of treatment with immunosuppression in these patients are unclear. Previous studies evaluating treatment with corticosteroids or immunosuppression have not demonstrated a change in the natural history of fibrotic lung disease [Paramothayan et al. 2005, 2006]. However, treatment of concomitant active inflammation may have a clinical benefit with respect to improvement in symptoms. As such, a brief trial of corticosteroids in patients with significant pulmonary symptoms and fibrotic lung disease may be justified.
Lung transplant is a potential lifesaving option for patients with end-stage lung disease due to sarcoidosis, and accounts for 2.5% of all lung transplants [Christie et al. 2012]. The optimal timing for referral is difficult to define. This is largely due to the chronic and variable natural history of the disease [Orens et al. 2006]. Current International Society for Heart and Lung Transplant (ISHLT) guidelines recommend referral for transplant evaluation in patients with New York Heart Association functional class III or IV impairment [Orens et al. 2006]. While guidelines for transplant include functional impairment, hypoxemia at rest, PH, and right atrial pressures exceeding 15 mmHg [Orens et al. 2006]. Elevated right atrial pressure is a particularly poor prognostic sign and is associated with increased mortality [Arcasoy et al. 2001]. Additional variables portending a worse prognosis while awaiting transplant include hypoxemia, elevated pulmonary artery pressure, and low cardiac output [Arcasoy et al. 2001].
Candidacy for lung transplant considers both the severity of lung disease and the risk of surgery. The presence of mycetomas, pleural disease, bulky hilar lymphadenopathy, and perihilar fibrosis predict technical difficulties with the surgery itself and should be taken into consideration [Shlobin and Nathan, 2012]. The presence of extrapulmonary disease weighs into the patient risk profile as well. As with all other lung transplant candidates, the patient must have adequate social support, be compliant with medical treatments, and should have manageable comorbid conditions. Therefore, the decision to transplant should be individualized. Survival after transplant is comparable to that of other lung diseases. Survival rates at 1, 5, and 10 years are 72.2%, 50.6%, and 31.1% respectively [Hertz et al. 2011; Shlobin and Nathan, 2012]. Recurrence of disease after transplant has been reported.
Complications of cavitary lung disease
Cavitary lesions are commonly seen in patients with fibrotic pulmonary sarcoidosis. Cavities can be bilateral or unilateral, and like other radiographic abnormalities in pulmonary sarcoidosis, they tend to occur in the upper lobes. Approximately 75% do not resolve and become permanent structural changes [Hours et al. 2008]. Persistent cavities predispose patients to formation of mycetomas, which have an incidence of 2% [Pena et al. 2011]. Mycetomas are most often caused by Aspergillus species. However, a variety of other fungal species can be involved. Mycetomas are most often found incidentally or after an episode of hemoptysis. Hemoptysis is proposed to be a result of several potential mechanisms, including local friction of a mobile fungus ball against a hypervascular cavity wall, invasion of Aspergillus into the surrounding vasculature, or local tissue damage relating to toxin and enzyme release by the fungus. Approximately 90% of patients with mycetomas experience at least one episode of hemoptysis [Patterson and Strek, 2013]. The spectrum of severity ranges from a mild self-limited episode to a massive life-threatening event.
For significant hemoptysis, angiography and selective bronchial artery embolization (BAE) is indicated to control bleeding. In a study of 12 patients with massive hemoptysis due to pulmonary aspergilloma, embolization of systemic and bronchial arteries resulted in resolution of hemoptysis within 24 h [Corr, 2006]. None of these patients had recurrence over the next 4 weeks [Corr, 2006]. Although this approach is less frequently utilized, the intracavitary instillation of amphotericin B, voriconazole, or potassium iodide solution has been shown to effectively treat mycetoma-related hemoptysis in nonsurgical candidates [Kravitz et al. 2011, 2013; Rumbak et al. 1996]. This approach is shown to be well tolerated and frequently results in sustained (i.e. 1–70 months) resolution of hemoptysis within 72 h [Kravitz et al. 2013; Rumbak et al. 1996]. Surgical resection is curative and can be lifesaving in cases of hemoptysis refractory to BAE or other conservative measures. Surgery is often complex and is associated with frequent complications, including postoperative hemorrhage, bronchopleural fistulas, wound infection, and empyema [Brik et al. 2008; Guerra et al. 2008]. Additionally, extensive anatomic involvement and poor preoperative pulmonary function may result in permanent respiratory impairment if surgery is undertaken. As such, resection is only possible in select patients and decisions relating to this procedure should be made in collaboration with an experienced thoracic surgeon.
Current Infectious Disease Society of America guidelines recommend systemic antifungal therapy for chronic cavitary pulmonary aspergillosis [Walsh et al. 2008]. Chronic cavitary pulmonary aspergillosis is defined as Aspergillus growth from respiratory cultures in the presence of multiple cavities (with or without aspergilloma) associated with pulmonary and systemic symptoms and elevated serum inflammatory markers. Lon-term therapy with systemic azoles may provide improvement in symptoms and stabilization of radiographic abnormalities in these patients [Walsh et al. 2008]. Voriconazole is first-line therapy. Itraconazole and posaconazole have also been used with good results. Systemic antifungals are not required for isolated aspergilloma, for which surgical resection is the definitive treatment. The benefits of surgical resection should be weighed against the risks of compromised pulmonary function and procedural complications. Treatment with antifungals prior to surgical intervention may help decrease the burden of fungal organisms and reduce the incidence of postsurgical infections [Patterson and Strek, 2013]. The same is true for antifungal therapy prior to lung transplant [Shlobin and Nathan, 2012]. However, the data to support their use are limited and should be considered on a case by case basis. In our practice, azoles are also used during treatment with immune-suppressing agents, which is often necessary to prevent further progression of associated pulmonary and systemic sarcoidosis.
Pulmonary hypertension
PH occurs in 5–6% of patients with sarcoidosis and is a daunting complication of the disease [Handa et al. 2006; Nunes et al. 2012]. Sarcoidosis-associated PH most commonly occurs in those with advanced lung disease. However, it can also present in the absence of fibrosis by a number of different mechanisms [Diaz-Guzman et al. 2008; Nunes et al. 2012]. The signs and symptoms of sarcoidosis-associated PH parallel those of other causes. It should be considered in those with severe dyspnea, signs of heart failure on examination, or persistent dyspnea despite adequate treatment with systemic corticosteroids. Reduced DLCO out of proportion to changes in lung volumes is also suggestive of PH. Transthoracic echocardiography is the recommended screening test, but does not definitively exclude the diagnosis or accurately predict severity. Right heart catheterization is the gold standard method of diagnosis. It also allows for evaluation of left heart dysfunction and assessment of vasodilator response.
Development of PH portends a poor prognosis in patients with sarcoidosis, and accounts for significant morbidity and mortality [Preston et al. 2001]. Mortality rates among patients with sarcoidosis awaiting transplant are independently associated with elevated right atrial pressures [Arcasoy et al. 2001]. The presence of PH among patients with stage IV pulmonary disease was associated with an 8.1-fold increased risk of death in one study [Nardi et al. 2011]. In light of these data, ISHLT guidelines recommend early evaluation for transplant in patients with advanced lung disease due to sarcoidosis complicated by pulmonary hypertension [Orens et al. 2006].
In addition to early transplant evaluation, general treatment for sarcoidosis-associated PH includes supplemental oxygen, diuretic therapy, and standard treatment of any comorbidity that may contribute to right heart dysfunction. These include obstructive sleep apnea (OSA), cardiac issues, and thromboembolic disease. Patients with sarcoidosis are at increased risk of OSA, with an incidence of 17% in one study [Turner et al. 1997].
Treatment of sarcoidosis-associated PH with corticosteroids is not routinely effective. However, a 12-month follow up of 24 patients with nonfibrotic pulmonary sarcoidosis with PH showed that a significant fraction of the experienced improved pulmonary hemodynamics, supporting the notion that some cases of sarcoidosis-induced PH are responsive to immunomodulating treatments [Gluskowski et al. 1990]. Patients with active inflammation, extrinsic compression of the pulmonary vasculature by mediastinal or hilar lymphadenopathy, and those with sarcoidosis vasculopathy are more likely to benefit from treatment [Gluskowski et al. 1990; Nunes et al. 2006]. Conversely, those with fibrotic lung disease and PH tend to have no significant improvement in outcomes with corticosteroid treatment [Nunes et al. 2006]. Thus, a brief trial of steroids is justified in patients with PH in the absence of fibrotic lung disease.
The use of pulmonary vasodilators for the treatment of sarcoidosis-associated PH is controversial. Current data regarding the use of vasodilators in patients with sarcoidosis are limited. To date, there are no randomized controlled trials for these agents in patients with sarcoidosis. A meta-analysis by Dobarro and colleagues showed improvement in 6MWD and hemodynamics (mean pulmonary artery pressure and pulmonary vascular resistance) [Dobarro et al. 2013]. In addition, cohort studies have demonstrated objective improvements in functional class and quality of life with treatment [Barnett et al. 2009; Baughman et al. 2006b; Fisher et al. 2006; Preston et al. 2001]. Notably, no study has shown any benefit with respect to mortality. A summary of outcomes from studies evaluating vasodilators in sarcoidosis-associated PH is shown in Table 4. Overall, vasodilators are useful in select patients as a palliative measure or as a ‘bridge’ to transplant [Barnett et al. 2009]. The benefits of therapy must be weighed against the potential for adverse side effects, including worsening hypoxemia or dyspnea due to increased ventilation–perfusion mismatch, development of pulmonary edema similar to effects seen in pulmonary veno-occlusive disease, and drug toxicity. Therefore, initiation of vasodilator therapy in patients with sarcoidosis-associated PH should be performed with caution and only after careful evaluation. Ideally, this should be performed in consultation with a PH specialist.
Outcomes of studies: evaluation of vasodilators for sarcoidosis-associated pulmonary hypertension.
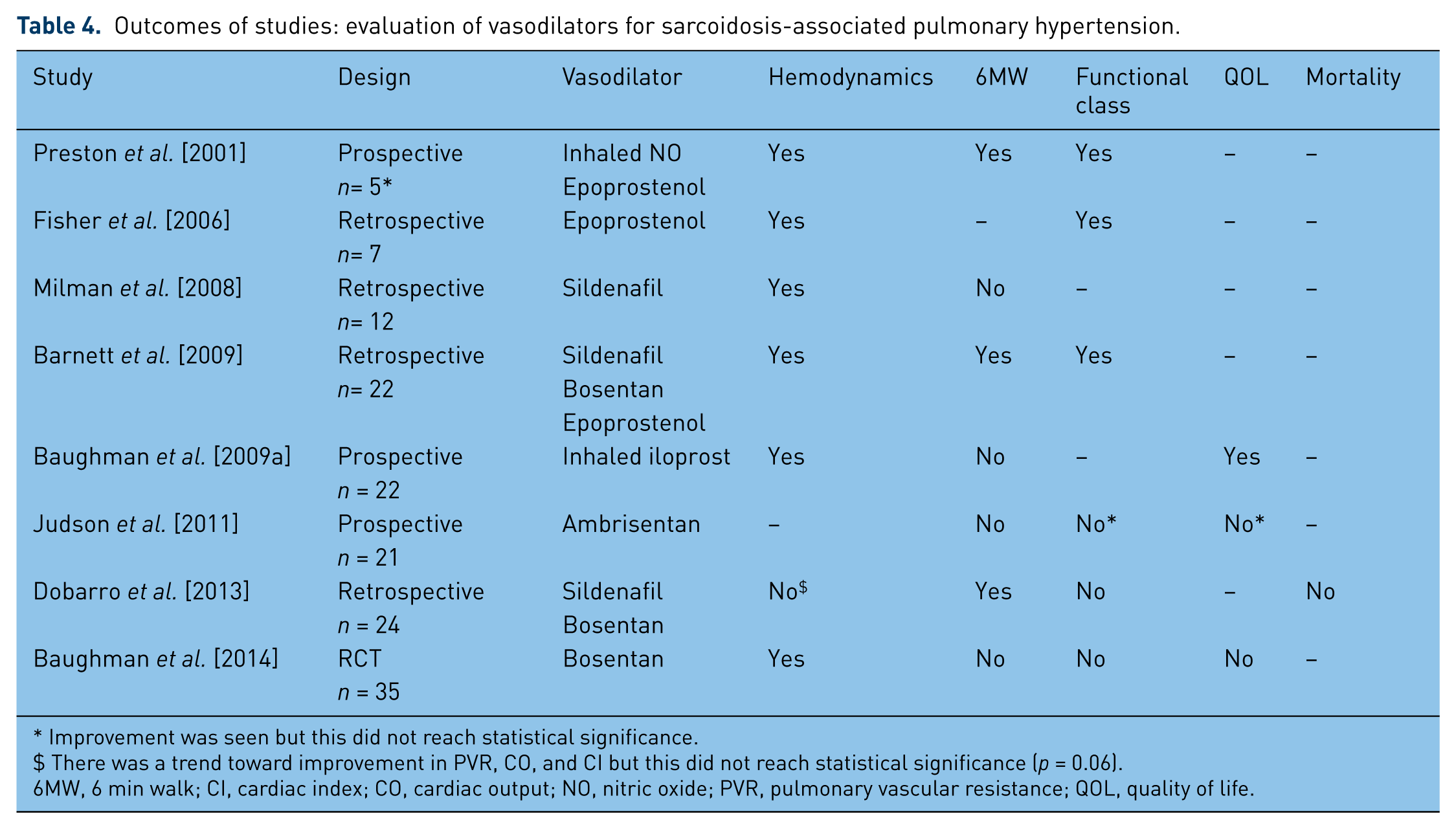
Improvement was seen but this did not reach statistical significance.
There was a trend toward improvement in PVR, CO, and CI but this did not reach statistical significance (p = 0.06).
6MW, 6 min walk; CI, cardiac index; CO, cardiac output; NO, nitric oxide; PVR, pulmonary vascular resistance; QOL, quality of life.
Cardiac sarcoidosis
CS is an increasingly recognized extrapulmonary manifestation that warrants special attention. The prevalence of cardiac involvement has been reported to range from 5% based upon clinical criteria to 39% or greater when using the latest imaging techniques [Judson et al. 2012; Mehta et al. 2008; Silverman et al. 1978]. Although many cases escape detection, CS is the second leading cause of sarcoidosis-related death in the USA and is the leading cause of death in Japanese patients [ATS/ERS/WASOG, 1999; Iwai et al. 1994]. The manifestations vary and can be clinically silent or manifest as heart failure, arrhythmias, or sudden cardiac death [Nunes et al. 2010]. The diagnosis of CS remains challenging, as reflected by the fact that many patients are first diagnosed with CS at the time of autopsy [Fabre and Sheppard, 2006; Matsui et al. 1976; Silverman et al. 1978]. Guidelines developed in 1999 recommend screening for myocardial involvement with a thorough history and ECG at initial diagnosis of sarcoidosis [ATS/ERS/WASOG, 1999]. However, this approach identifies a minority of affected patients. A single-center study demonstrated the sensitivity of detecting cardiac sarcoidosis to be greatly enhanced by combining screening modalities, including history, ECG, 24 h Holter monitor, and transthoracic echocardiogram [Mehta et al. 2008]. There is reason to believe that more advanced imaging with cardiac MRI or PET scan may further improve the detection of CS [Crouser et al. 2014]. Endomyocardial biopsy may be helpful if positive but lacks sensitivity due to the patchy distribution of the granulomas and should not be routinely performed [Bussinguer et al. 2012; Nunes et al. 2010]. The Japanese Ministry of Health and Welfare developed guidelines for the diagnosis of CS in 1993, as seen in Table 5 [Hiraga et al. 1993]. These guidelines were updated in 2006 to consider the role of more sophisticated imaging modalities such as cardiac MRI [Soejima and Yada, 2009]. These guidelines are due for additional modifications based upon emerging data relating to the utility of noninvasive imaging for diagnosis and risk stratification (e.g. predicting potentially fatal arrhythmias) [Crouser et al. 2014].
Guidelines for the diagnosis of cardiac sarcoidosis established by the Japanese Ministry of Health and Welfare, 1993 [Hiraga et al. 1993].

The diagnosis of CS warrants strong consideration for treatment with systemic corticosteroids. Similar to pulmonary disease, damage to the heart can be caused by acute inflammation, fibrosis, or both [Yang et al. 2014]. Furthermore, it is thought that potentially reversible inflammation evolves into irreversible inflammation. As such, suppression of active cardiac inflammation seems prudent, and higher doses of prednisone in the range of 60–80 mg/day during the initial phase of treatment were previously recommended [Bussinguer et al. 2012]. However, objective evidence indicates no advantage of using more than 30 mg/day prednisone or equivalent [Yazaki et al. 2001], which is consistent with our practice patterns. Given the long-term complications associated with even moderate doses of prednisone (e.g. >10 mg/day), we advocate for the use of a steroid-sparing agent to facilitate the tapering of prednisone to 10 mg/day or less, as tolerated based upon clinical response. The optimal duration of treatment is unknown and may vary on a case by case basis. However, it is unclear how to best monitor disease progression. Patients presenting with arrhythmias should be referred for electrophysiologic evaluation with consideration of implantable cardioverter defibrillator (ICD) placement. While the American Heart Association recommends (level IIB) that all patients with CS have an ICD placed, this is impractical. Up to 40% of patients will have CS if carefully assessed using the most current imaging techniques [Greulich et al. 2013], and only a fraction of these patients will develop clinically significant cardiac complications [Mehta et al. 2008]. The high costs and potential complications (e.g. lead displacement, inappropriate deployment, infections) associated with ICD placement may not justify placement in all patients. There are certain high-risk features that strongly favor ICD placement and other features that are associated with low risk [Aizer et al. 2005; Mehta et al. 2008]. There is a pressing need for further studies to guide clinicians in the management of CS. Our evaluation of patients with cardiac sarcoidosis has been previously published and is shown in Figure 3 [Evanchan et al. 2013].

Proposed algorithm for the treatment of cardiac sarcoid. Permission granted for reprint from Evanchan et al. [2013]. ECG, electrocardiogram; EF, ejection fraction; ICD, implantable cardioverter defibrillator; MRI, magnetic resonance imaging; PET, positron emission tomography.
Small fiber neuropathy
Small fiber neuropathy (SFN) is now appreciated as a common complication of sarcoidosis. It has been noted that over 40% of patients with sarcoidosis have symptoms of SFN, which include peripheral pain, paresthesias, or autonomic dysfunction [Hoitsma et al. 2002]. The vague manifestations of SFN often obscure the diagnosis. In our experience, many such patients are misclassified as having fibromyalgia. A clinical screening questionnaire has been developed as a tool to help identify patients with symptoms consistent with SFN [Hoitsma et al. 2011]. Electromyography and nerve conduction testing are normal in patients with SFN and other testing is required to make the diagnosis. Skin biopsy is an accurate way to quantify intraepithelial nerve fiber density. The diagnosis of SFN is supported by the finding of lower than normal nerve fiber density [Tavee and Culver, 2011; Tavee and Zhou, 2009]. More recently, corneal confocal microscopy has emerged as a noninvasive way to measure small fiber density and length. It has been shown that this method also correlates with severity of neuropathy experienced by the patient [van Velzen et al. 2014]. However, corneal confocal microscopy is not readily available at most clinical centers. Autonomic testing with quantitative sudomotor axon reflex testing is more commonly used to evaluate for SFN. This is performed by measuring sweat output in response to stimulation and is compared with normal values [Tavee and Culver, 2011; Tavee and Zhou, 2011]. Once the diagnosis has been made, treatment should be considered to improve the patient’s often debilitating symptoms. Traditional treatment with steroids is not beneficial and other immunomodulating drugs have been explored. Intravenous immunoglobulin improved SFN symptoms in a small case series [Parambil et al. 2011]. Anti-TNFα agents also show promise [Elfferich et al. 2010; Parambil et al. 2011]. Most recently, an anti-inflammatory drug, ARA 290, has been demonstrated to be safe and effective in the treatment of SFN in patients with sarcoidosis in the Netherlands [Heij et al. 2012]. The role of exercise and other lifestyle modifications are undefined at this time and many patients are physically limited due to chronic pain.
Fatigue
Sarcoidosis-associated fatigue is a common and problematic symptom to manage. It has been found that 70–83% of patients with sarcoidosis experience fatigue [de Kleijn et al. 2009; Hinz et al. 2011]. The Fatigue Assessment in Sarcoidosis (FAS) scale, a validated 10-item questionnaire, has served as a useful tool to more objectively quantify severity. It may assist in the diagnosis and management of patients with this debilitating symptom [De Vries et al. 2004]. The etiology of fatigue in the sarcoidosis population is likely multifactorial and has been associated with sleep-related disorders, depression, metabolic abnormalities, SFN, drug-related, and inflammatory etiologies [Drent et al. 2012]. The initial evaluation of fatigue should focus on identifying and correcting these potentially treatable conditions. Despite adequate end-organ treatment response to therapy, many patients with sarcoidosis experience persistent fatigue [Korenromp et al. 2011]. In such cases, limited data support the use of neurostimulants or TNFα inhibitors. A small (10 subject) prospective randomized crossover study of dexmethylphenidate showed a significant improvement in fatigue symptoms, as measured by the FAS scale [Lower et al. 2008]. Likewise, armofafinil was shown to be beneficial in a small, prospective, randomized crossover study of similar design [Lower et al. 2013]. Therapy with the TNFα inhibitors may also improve fatigue in patients with sarcoidosis. This was demonstrated in a study that showed a significant improvement in FAS scores in patients treated with adalimumab and infliximab compared with patients not treated or treated with prednisone with or without methotrexate [Elfferich et al. 2010]. The role of exercise therapy to improve fatigue is unclear but warrants further study.
Calcium and vitamin D metabolism
Management of calcium and vitamin D metabolism in patients with sarcoidosis presents an important and difficult challenge. Large epidemiologic studies have shown that a minority of patients with sarcoidosis have overt disorders of calcium and vitamin D homeostasis [Baughman et al. 2001]. However, adverse effects on bone health, particularly osteoporosis and fractures, are very common in patients with sarcoidosis. There is evidence that up to half of patients with untreated sarcoidosis develop osteopenia [Rizzato, 1998]. The risk of bone complications further increases in the setting of chronic treatment drugs affecting calcium metabolism, including glucocorticoids and methotrexate. Studies suggest the use of glucocorticoids is a risk factor for fracture independent of bone density [Grossman et al. 2010; Kanis et al. 2004b]. The American College of Rheumatology (ACR) has recently updated its guidelines concerning the appropriate prevention of glucocorticoid-induced osteoporosis in 2010 [Grossman et al. 2010]. While these recommendations are not specific for sarcoidosis, many of the recommendations are readily applied. The ACR guidelines use the World Health Organization Fracture Assessment Risk model to help risk stratify patients into low-, medium-, and high-risk groups. In low-risk patients, the ACR recommends the use of a bisphosphonates to prevent osteoporosis in postmenopausal woman or men older than 50 years if they are receiving the equivalent of 7.5 mg of prednisone or higher per day [Grossman et al. 2010]. In premenopausal women and men under 50 years old, the recommendations are less clear. Medium- and high-risk patients should be considered for bisphosphonate therapy at any daily dose of glucocorticoid [Grossman et al. 2010]. Calcitonin and the bisphosphonate, alendronate, have both been shown to be effective at preventing corticosteroid-induced osteoporosis in patients with sarcoidosis [Gonnelli et al. 1997; Montemurro et al. 1991].
The ACR strongly recommends calcium and vitamin D supplementation for anyone treated long term with corticosteroids [Grossman et al. 2010]. In patients with sarcoidosis, it is unlikely that this recommendation can be applied broadly without consideration. There are several case reports of patients with sarcoidosis developing hypercalcemia after receiving high-dose vitamin D replacement [Amrein et al. 2011]. Patients with sarcoidosis have been shown to have low levels of 25-OH vitamin D but the majority will have normal levels of activated 1,25-OH vitamin D. Experts have cautioned against supplementing with high-dose vitamin D in this situation [Burke et al. 2010; Sweiss et al. 2011]. However, lower doses of vitamin D for osteoporosis prevention are likely to be safe in patients without hypercalcemia or hypercalcemia [Conron et al. 2000]. Close monitoring of calcium and vitamin D levels should be performed if supplementation is given [ATS/ERS/WASOG, 1999; Conron et al. 2000].
Summary
The treatment of pulmonary sarcoidosis is rapidly evolving in parallel with improved diagnostic testing (e.g. improved imaging for cardiac sarcoidosis) and the availability of alternative treatment options. Given that sarcoidosis is among the most common chronic interstitial lung diseases and commonly afflicts young to middle-aged adults, there is a pressing need to further improve the detection and treatment of sarcoidosis. It is further apparent from this lengthy review that the management of pulmonary disease is challenging in that uncertainty remains with respect to the optimal treatment protocols and the systemic nature of the disease often requires effective collaboration among a team of experienced specialists.
Footnotes
Acknowledgements
All authors contributed equally to the preparation of the manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.