
Abstract
Pulmonary hypertension (PH) is characterized by a progressive elevation of pulmonary arterial pressure due to alterations of both pulmonary vascular structure and function. This disease is rare but life-threatening, leading to the development of right heart failure. Current PH treatments, designed to target altered pulmonary vascular reactivity, include vasodilating prostanoids, phosphodiesterase-5 inhibitors and endothelin-1 receptor antagonists. Although managing to slow the progression of the disease, these molecules still do not cure PH. More effective treatments need to be developed, and novel therapeutic strategies, targeting in particular vascular remodelling, are currently under investigation. Reactive oxygen species (ROS) are important physiological messengers in vascular cells. In addition to atherosclerosis and other systemic vascular diseases, emerging evidence also support a role of ROS in PH pathogenesis. ROS production is increased in animal models of PH, associated with NADPH oxidases increased expression, in particular of several Nox enzymes thought to be the major source of ROS in the pulmonary vasculature. These increases have also been observed in vitro and in vivo in humans. Moreover, several studies have shown either the deleterious effect of agents promoting ROS generation on pulmonary vasculature or, conversely, the beneficial effect of antioxidant agents in animal models of PH. In these studies, ROS production has been directly linked to pulmonary vascular remodelling, endothelial dysfunction, altered vasoconstrictive responses, inflammation and modifications of the extracellular matrix, all important features of PH pathophysiology. Altogether, these findings indicate that ROS are interesting therapeutic targets in PH. Blockade of ROS-dependent signalling pathways, or disruption of sources of ROS in the pulmonary vasculature, targeting in particular Nox enzymes, represent promising new therapeutic strategies in this disease.
Keywords
Introduction
Pulmonary hypertension (PH) is a rare and severe disease, with a progressive elevation of pulmonary arterial pressure (PAP) often leading to right heart failure. Therapeutic strategies currently available, mostly designed to induce vasodilation, alleviate PH symptoms but do not afford a cure. Clinical worsening leads to a necessary heart/lung graft with reduced life expectancy. PH cellular and molecular mechanisms are therefore under investigation in order to define new therapeutic targets. Among the new potential strategies that have emerged recently, targeting of reactive oxygen species (ROS) appears as an interesting approach in PH treatment. Indeed, ROS play important physiological roles in the pulmonary vasculature, and recent studies have also evidenced a role for ROS in PH pathophysiology.
After a brief summary of PH pathophysiology and treatments currently available, this review will provide information about ROS production and metabolism in the pulmonary vasculature, ROS-dependent signalling pathways in pulmonary vascular cells and will summarize the data showing a role for ROS in the pathogenesis of PH.
Pathophysiology of PH
PH is defined as an elevation of mean PAP above 25 mmHg at rest, compared with normal mean PAP comprised between 10 and 15 mmHg [Simonneau et al. 2009]. PH is characterized by both functional and structural modifications of pulmonary arteries, leading to altered pulmonary vascular reactivity and remodelling of the pulmonary arterial wall [Schermuly et al. 2011]. These mechanisms are associated with thrombotic lesions of pulmonary arteries [Humbert et al. 2004; Voelkel et al. 2012]. Increased pulmonary vascular resistance leads to increase in right heart work to maintain cardiac output, ultimately leading to right heart failure and death. PH classification has been updated recently and PH forms are now divided into five categories: pulmonary arterial hypertension belongs to group 1, being idiopathic, familial or associated with other diseases including for example connective tissue diseases, HIV infection, congenital heart disease or portal hypertension. Group 2 includes PH caused by left-sided heart disease, such as for example cardiomyopathy, diastolic dysfunction or aorticstenosis. Secondary PH forms associated with chronic respiratory diseases such as chronic obstructive pulmonary diseases or pulmonary fibrosis are often named ‘hypoxic’ PH and belong to group 3. Group 4 includes PH due to pulmonary embolus or pulmonary thrombosis. Group 5 includes PH due to other miscellaneous causes, which do not fit into the other four categories [Simonneau et al. 2009].
Altered pulmonary vascular reactivity evidenced in PH is characterized by an increased contraction associated with a decreased endothelium-dependent relaxation of pulmonary arteries. This phenomenon is due in particular to an endothelial dysfunction leading to an imbalance in the production/bioactivity of both vasoconstrictive mediators such as serotonin or endothelin-1 and vasodilators such as nitric oxide (NO) or prostacyclin (PGI2) [Humbert et al. 2004]. The function of the pulmonary arterial smooth muscle is also modified, with altered contractile signalling pathways, such as the RhoA/ROCK pathway [Connolly and Aaronson, 2011], and decreased expression/activity of ions channels, such as voltage-gated potassium channels [Guibert et al. 2007; Rhodes et al. 2009], leading to excessive contraction of pulmonary arteries.
Structural changes observed in pulmonary arteries, also called pulmonary vascular remodelling, include medial and adventitial thickening in proximal arteries, due to decreased apoptosis, hyperplasia and hypertrophy of smooth muscle cells and of fibroblasts associated with excessive accumulation of matrix proteins [Morrell et al. 2009]. Formation of a neointima is also observed, due to accumulation of smooth muscle cells and myofibroblasts, together with the formation of plexiform lesions resulting from excessive and disorganized smooth muscle and endothelial cell proliferation. In normally nonmuscular distal arteries, formation of a media de novo is observed, resulting from recruitment, differentiation and proliferation of fibroblasts and progenitor cells [Hassoun et al. 2009; Schermuly et al. 2011]. All of these structural changes are therefore leading to arterial wall thickening, thus contributing to the increased pulmonary vascular resistance and elevation of PAP observed in PH.
Current treatments of PH
The current therapeutic strategy developed for PH is to associate a ‘specific’ treatment, against excessive pulmonary vascular contraction and remodelling, to a more ‘conventional’ treatment dedicated to alleviate symptoms, effort and right heart failure [Frumkin, 2012; O’Callaghan et al. 2011]. Conventional treatments comprise lifestyle modification, diuretics to alleviate right heart failure, anticoagulant therapies to reduce risks of thrombotic lesions and oxygen therapy to treat hypoxemia [Galie et al. 2004; Simonneau et al. 2009]. Whereas nonselective systemic vasodilators such as calcium channel blockers are effective only in a few groups of responding patients [Montani et al. 2010; Rubin, 1985; Tonelli et al. 2010], more specific vasodilator treatments have been developed for the treatment of PH. Three families of molecules are currently used: vasodilating prostanoids, phosphodiesterase-5 inhibitors and endothelin-1 receptor antagonists [Frumkin, 2012].
Prostacyclin, also named prostaglandin I2 (PGI2), is a potent vasodilator whose endothelial production is decreased in PH. This decrease therefore contributes to excessive pulmonary arterial contraction, but also to pulmonary vascular remodelling, since PGI2 normally also inhibits smooth muscle proliferation, platelet aggregation and exerts anti-inflammatory actions [Gomberg-Maitlandand Olschewski, 2008]. Prostacyclin therapy has therefore been employed for over 25 years to treat PH and showed efficiency in alleviating symptoms, and even in increasing patient survival for epoprostenol [Sitbon et al. 2002]. However, because of its complex intravenous administration and its numerous adverse effects, more stable prostacyclin analogues have been developed with other routes of administration, in particular orally, subcutaneously or inhaled active compounds [Yildiz, 2009]. Therapy strategies combining oral prostacyclin analogues with phosphodiesterase-5 inhibitors and/or endothelin-1 receptor antagonists have also been developed to enhance therapeutic efficiency.
Endothelin-1, activating ETA and ETB receptors, is a potent vasoconstrictor and also participates in smooth muscle proliferation, fibrosis and inflammation [Kim and Rubin, 2002]. Activation of ETA receptors, mainly expressed on smooth muscle cells, induces vasoconstriction whereas activation of ETB receptors, also expressed on endothelial cells, induces NO release and vasodilation [Dupuis, 2001]. However, both ETA selective antagonists such as ambrisentan and dual ETA/ETB antagonists such as bosentan are used in clinics and have all been shown to alleviate PH symptoms and to improve exercise capacity [Galie et al. 2008a, 2008b; O’Callaghan et al. 2011; Rubin et al. 2002].
NO-mediated relaxation of the pulmonary vascular smooth muscle involves synthesis of cyclic guanosine monophosphate (cGMP), which is then degraded by phosphodiesterase-5 (PDE5). Inhibitors of PDE5 such as sildenafil and tadalafil are therefore also used in PH treatment. PDE5 is abundantly expressed in the pulmonary vasculature [Hanson et al. 1998; Pauvert et al. 2003] and PDE5 inhibitors induce beneficial pulmonary vasodilation and antiproliferative effects in PH patients [Archer and Michelakis, 2009; Montani et al. 2009].
In summary, the great progress made in the comprehension of PH pathophysiological mechanisms has led to the development of these specific treatments and greatly improved patient’s clinical course and life expectancy. However, although being able to alleviate PH symptoms, these treatments still do not afford a cure, and this shows the need to persevere in the comprehension of PH cellular and molecular mechanisms in order to define new therapeutic strategies. Among the various targets under investigation at the moment, targeting of ROS appears as an interesting new approach in PH treatment.
Targeting ROS as a novel therapeutic strategy in PH
ROS are cellular products derived from oxygen metabolism. Depending on their local concentration, ROS can play physiological but also pathophysiological roles, becoming harmful to cells. Indeed, at low concentrations, ROS are intracellular signalling messengers, and ROS production is counterbalanced by antioxidant systems contributing to ROS metabolism [Touyz, 2004]. However, an imbalance between ROS production and ROS metabolism can lead to increased ROS levels, a situation named oxidative stress, with alteration by oxidation of essential cellular components such as lipids, proteins, mitochondria and even DNA, which can ultimately lead to cell death.
The vasculature is an important source of ROS [Touyz, 2004], and many studies have shown a role for ROS in vascular physiology and pathophysiology. Indeed, oxidative stress is a mediator of vascular injury in cardiovascular diseases such as systemic hypertension [Touyz, 2004], atherosclerosis [Victor et al. 2009] or ischemia–reperfusion [Murphy and Steenbergen, 2008]. A growing body of evidence, summarized in the following sections, also demonstrates a role for ROS and for oxidative stress in various aspects of PH pathophysiology.
Production and metabolism of ROS in the pulmonary vasculature
Biology of ROS
Many products are generated in normal cell respiration, derived from the reduction of molecular oxygen and all termed ROS. However, ROS have different chemical properties, being either free or nonfree radicals. Indeed, some ROS have an unpaired electron on their outer orbital, such as superoxide (
ROS generation often begins with one- or two-electron reduction of molecular oxygen to form

Generation and metabolism of ROS.
Sources of ROS in the pulmonary vasculature
In mammalian cells, enzymatic sources of ROS include NADPH oxidases, uncoupled nitric oxide synthase (NOS), cytochromes P450, xanthine oxidase (XO), the mitochondrial electron transport chain, and other enzymes such as lipoxygenases, cyclooxygenases or peroxidases as well as hemoproteins such as heme and hematin [Griendling et al. 2000]. All vascular cell types are able to produce ROS either from metabolic or from enzymatic sources. However, in the pulmonary vasculature, ROS main sources are NADPH oxidases, uncoupled endothelial NOS (eNOS), XO as well as the mitochondrial electron transport chain [Li and Shah, 2004; Lyle and Griendling, 2006].
NAPDH oxidases
NADPH oxidases are enzymes composed of multiple subunits that catalyse
The mammalian Nox family comprises seven members: Nox1, Nox2, Nox3, Nox4, Nox5, Duox1 and Duox2 [Brandes et al. 2010]. They all differ in their mode of activation, expression, and/or interaction with other proteins. In the cardiovascular system, Nox1, Nox2, Nox4 and Nox5 have been detected [Brandes et al. 2010]. The prototypical phagocytic NADPH oxidase (Nox2) is composed of five subunits (Figure 2): the two membrane-bound subunits gp91phox (the catalytic subunit, also termed Nox2) and p22phox, forming a heterodimeric complex named cytochrome b558, and the three cytosolic subunits p40phox, p47phox and p67phox. In stimulated cells, p47phox phosphorylation leads to assembly of the three cytosolic subunits to form a complex that translocates to the membrane and associates with cytochrome b558 to assemble the active oxidase [Touyz and Briones, 2010]. This activation also requires the small G proteins Rac (Rac1 or Rac2) and Rap1A [Bokoch and Knaus, 2003]. Nox1 is the closest homolog of Nox2, and requires the membrane subunit p22phox as well as at least two other cytosolic subunits termed Noxo1 (Nox organizer 1, which is a p47phox analogue) and Noxa1 (Nox activator 1, which is a p67phox analogue) (Figure 2). Nox4 was initially termed Renox (for renal oxidase, as its expression is abundant in the kidney). Nox4 differs from Nox1 and Nox2 in the fact that it mainly produces H2O2 directly, whereas Nox1 and Nox2 first produce

Structure of NADPH oxidases expressed in pulmonary arteries.
In the lung, expression of the membrane-bound subunits Nox1, Nox2, Nox4 and p22phox have been shown in pulmonary vessels, together with expression of the cytosolic subunits Noxa1, Noxo1, p40phox, p47phox and p67phox [Perez-Vizcaino et al. 2010] (Figures 2 and 3). In cultured cells, expression of Nox2 and Nox4 has been reported in pulmonary arterial endothelial cells (PAECs) [Bayraktutan et al. 2000; Griffith et al. 2009], in adventitial fibroblasts [Haurani and Pagano, 2007], and in pulmonary arterial smooth muscle cells (PASMCs) [Brandes and Kreuzer, 2005; Mittal et al. 2007]. In all of these cells, Nox4 is the predominant Nox isoform expressed [Perez-Vizcaino et al. 2010]. Nox1 expression seems to be restricted to PASMCs and is present in low concentrations in basal states of these cells [Mittal et al. 2007] (Figure 3).

Molecular sources of ROS in pulmonary arteries.
Uncoupled endothelial NOS
NOS is the enzyme responsible for NO synthesis. Three isoforms have been identified: NOS1 (neuronal NOS or nNOS), NOS2 (inducible NOS or iNOS) and NOS3 (endothelial NOS or eNOS) [Zuckerbraun et al. 2011]. Active eNOS is a homodimer and catalyses the formation of NO from L-arginine. Each monomer contains a carboxy-terminal reductase domain and an amino-terminal oxygenase domain. The reductase domain is composed of two flavinic cofactors (flavin mononucleotide [FMN] and flavin adenine dinucleotide [FAD]) binding sites and also contains a NADPH binding site. The oxygenase domain contains binding sites for heme, for the substrate L-arginine and for the cofactor tetrahydrobiopterin (BH4). Oxygenase and reductase domains are linked with a small sequence of amino acids that contains a calmodulin binding site [Alderton et al. 2001]. Heme groups are essential for dimerization of both monomers and for promoting electron transfer from the flavin to the heme of the other monomer. Finally, a zinc ion binds at both monomers to stabilize the dimeric structure [Weseler and Bast, 2010]. An electron transfer is catalysed within the reductase domain via the flavins FMN and FAD to the heme in the oxygenase domain. Binding of the calcium-calmodulin complex to the reductase domain facilitates this electron transfer. O2 is then reduced at the heme of the oxygenase domain and L-arginine is oxidized to L-citrulline and NO●. Many signals can regulate eNOS activity, in particular intracellular calcium concentration or phosphorylation in the reductase and calmodulin-binding domains through the action of kinases such as protein-kinase B (PKB, also named Akt), protein-kinase A (PKA) or calmodulin-dependent kinase II (CamKII) [Fleming, 2010; Forstermann, 2006].
Under some conditions, eNOS generates
Xanthine oxidase
XO and xanthine dehydrogenase (XDH) are xanthine oxidoreductases (XORs), and are involved in the last steps of purine metabolism, catalysing the transformation of hypoxanthine and xanthine to uric acid [Jarasch et al. 1981]. XO and XDH are encoded by the same gene, and can be interconverted in vivo [Nishino et al. 2008]. XDH uses NAD+ as an electron acceptor, thus producing NADH, whereas XO prefers O2 as an electron acceptor, therefore generating
XO is composed of two monomers, each containing a molybdopterin moiety that catalyses oxidation of a substrate. The accepted electrons are then rapidly transported via a Fe2–S2 centre to the cofactor FAD, which then reduces the oxidant substrate O2 [Harrison, 2002]. Reoxidation of XO needs the transfer of six electrons to O2, therefore generating two molecules of H2O2 and two molecules of
Mitochondrial electron transport chain
Mitochondria are also a major source of ROS in the pulmonary vasculature. The mitochondrial matrix contains enzymes that participate in the metabolism of pyruvate and fatty acids to produce acetyl CoA. The matrix also contains enzymes of the citric acid cycle which can then oxidize acetyl CoA to CO2, generating three molecules of NADH and one molecule of FADH2 during this process. NADH and FADH2 are then used as electron donors by the mitochondrial electron transport chain to generate the proton gradient necessary to produce ATP.
The mitochondrial respiratory chain is the main energy source for the cell. Indeed, more than 95% of O2 consumed by cells is reduced by four electrons to yield two molecules of H2O. This occurs at the inner mitochondrial membrane, via mitochondrial electron transport chain complexes I to IV [Birukov, 2009; Fuchs et al. 2010] (Figure 3). NADH is oxidized by complex I, and FADH2 is oxidized by complex II, leading to reduction of ubiquinone to ubiquinol. Two electrons are carried by ubiquinol from complex I to complex III (coenzyme Q: cytochromes c-oxidoreductase). Another pair of electrons is transferred to ubiquinone at complex II, and is then also carried by ubiquinol to complex III. In complex III, one electron is transferred at the Q0 site to the Rieske iron–sulfur protein and then to cytochrome c, while the other electron is transferred to cytochrome b, followed by a transfer to ubiquinone at the Q0 site. Cytochrome c then donates electrons to O2 in complex IV, thus producing H2O [Shimoda and Undem, 2010].
Under physiological conditions, approximately 2–3% of the oxygen consumed by mitochondria is incompletely reduced, and mitochondria therefore also produce
In the pulmonary vasculature, production of ROS by mitochondria is regulated by modulation ofmitochondria biogenesis and bioenergetics [Erusalimsky and Moncada, 2007]. Modulation of these processes are dependent of multiple factors, including for example the availability of electron donors or the activity of cytokines and vasoactive mediators such as NO, angiotensin II or thromboxane A2 [Bailey et al. 2005; Rathore et al. 2006; Waypa et al. 2010]. In addition, a ROS-induced ROS release mechanism may also occur in the pulmonary vasculature, with for example angiotensin II and hypoxia triggering an increased ROS production after Nox activation in PASMC, leading to a subsequent increase in mitochondrial ROS production [Rathore et al. 2008]. Finally, the concentration of oxygen in pulmonary tissues is also particularly important in the modulation of ROS production by mitochondria, with ROS levels being elevated during hypoxia [Stowe and Camara, 2009].
Antioxidant systems in the pulmonary vasculature
Enzymatic and nonenzymatic antioxidant systems are present in vascular tissues to protect against deleterious oxidative stress. Pulmonary vascular nonenzymatic antioxidants can scavenge HO● and other free radicals and include ascorbate, glutathione, bilirubin, tocopherols and uric acid [Tajima et al. 2009]. Major pulmonary vascular enzymatic antioxidants include SOD, glutathione peroxidases, catalases, thioredoxin and peroxiredoxin [Touyz and Briones, 2010]. The major vascular SOD is the extracellular SOD that is produced and secreted by vascular smooth muscle cells and then binds to glycosaminoglycans in the vascular extracellular matrix [Gongora et al. 2006].
ROS-dependent signalling pathways in pulmonary vascular cells
ROS serve as important intracellular and intercellular messengers to regulate a variety of signalling pathways in pulmonary vascular cells. This redox signalling occurs mainly through reactions with protein residues, either with cysteine residues and formation of disulfide bridges, or through protein carbonylation [Perez-Vizcaino et al. 2010; Wong et al. 2008]. The main ROS targets in the pulmonary vasculature are described below and in Figure 4.
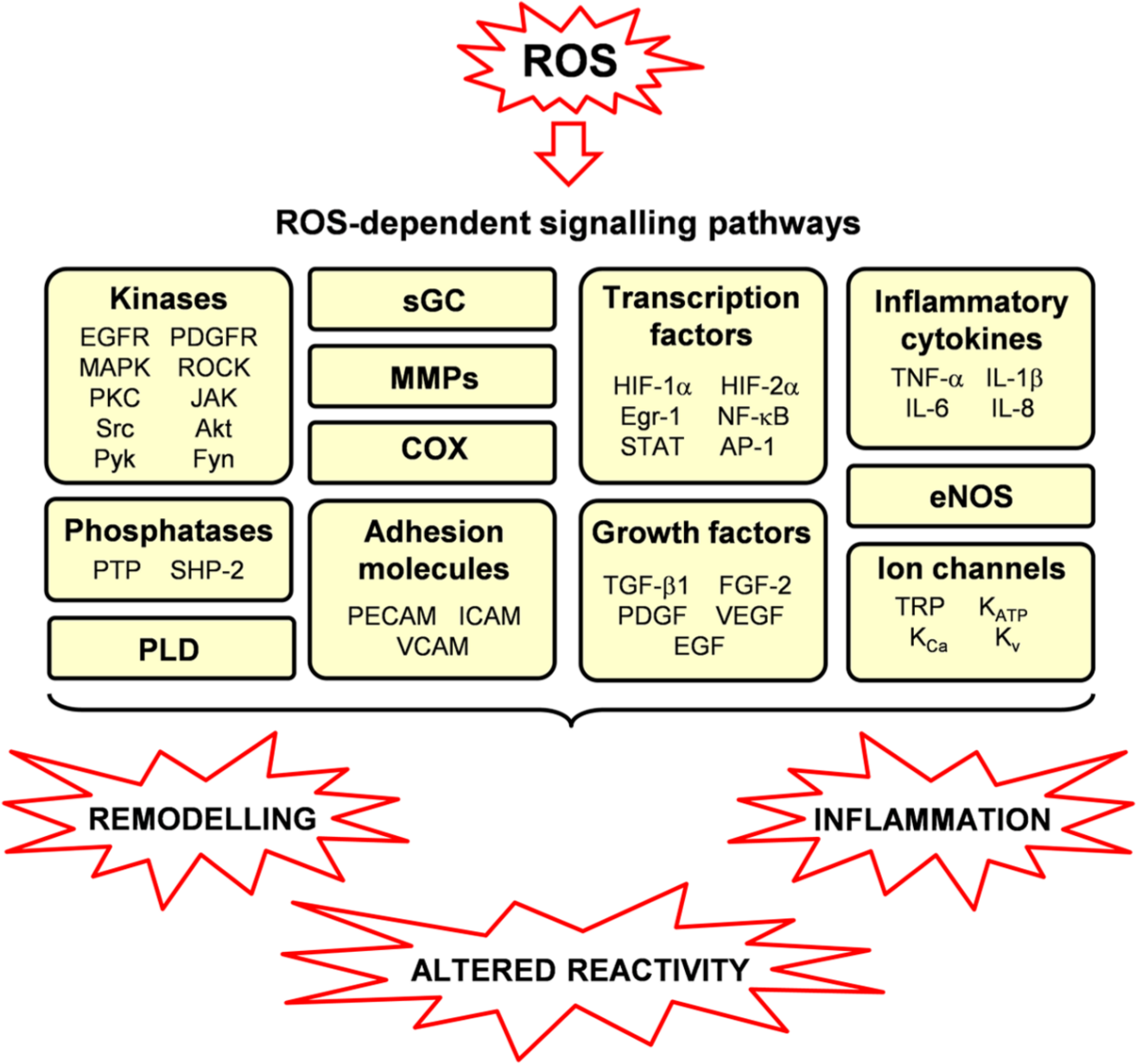
Redox-sensitive signalling pathways in pulmonary vascular cells and roles of ROS in pulmonary hypertension.
NO/guanylyl cyclase pathway
When superoxide production is increased, it can rapidly interact with NO and therefore attenuate its capacity to stimulate soluble guanylyl cyclase (sGC) in the systemic and pulmonary vasculatures [Perez-Vizcaino et al. 2010]. Moreover, oxidative stress can oxidize the ferrous (Fe2+)–heme and thiol sites on sGC which control its activation by NO, and therefore contribute to decreased cGMP production and smooth muscle relaxation [Wolin et al. 2010].
In addition, recent data obtained in our group show that ROS production in PASMC reduces endothelial NO-dependent control of pulmonary vasoreactivity, by targeting PAEC via myoendothelial gap junctions [Billaud et al. 2009]. This is the first direct evidence of a smooth muscle negative feedback on NO-dependent endothelial vasodilatation, through
Redox-sensitive enzymes
Receptor and nonreceptor tyrosine kinases
Classical activation of growth factor receptors by their agonist can be redox sensitive. For example, activation of its receptor by the platelet-derived growth factor (PDGF) is ROS-dependent [Sundaresan et al. 1995]. In addition, many studies have also focused on transactivation of growth factor receptors, which is a way of activating the receptor tyrosine kinase in the absence of any extracellular agonist [Knock and Ward, 2011]. This mechanism has been evidenced in particular in vascular smooth muscle cells for the epidermal growth factor receptor (EGFR) whose activation is induced by hydrogen peroxide or peroxynitrites in the absence of EGF [Knock and Ward, 2011; Ushio-Fukai et al. 2001]. Although these mechanisms have been evidenced in systemic vessels only, growth factors such as EGF or PDGF play an important role in particular in vascular remodelling observed in pulmonary arteries [Thomas, 2010]. Such ROS-dependent mechanism may also occur in the pulmonary vasculature and may therefore contribute to PH pathophysiology.
Among nonreceptor tyrosine kinases, the src family is of particular interest in the pulmonary vasculature [Knock and Ward, 2011]. Indeed, activation of Src kinase, in relation to activation of transcription factors such as the signal transducer and activator of transcription (STAT) 3 and the nuclear factor of activated T cells (NFAT), seems to play a pathophysiological role in PH [Courboulin et al. 2011; Paulin et al. 2011]. Since Src kinases have been implicated in regulation of hypoxia-induced contraction of pulmonary arteries [Knock et al. 2008], and since this mechanism seems to be, at least in part, mediated by ROS generation [Knock et al. 2008], the src kinase family may play a pivotal role in ROS-dependent signalling pathways in the pulmonary vasculature [Griendling et al. 2000; Knock and Ward, 2011], in both smooth muscle and endothelial cells. In the latter, the stimulatory effect of hydrogen peroxide on eNOS activity is src-dependent [Rafikov et al. 2011].
Protein tyrosine phosphatases
Protein tyrosine phosphorylation is one of the major post-transcriptional mechanisms and plays a critical role in many cell functions such as for example proliferation, differentiation and migration. The level of tyrosine phosphorylation is tightly regulated with a balance between protein tyrosine kinases (PTK) and protein tyrosine phosphatases (PTP) [Paravicini and Touyz, 2006]. All PTP possess a reactive and redox-regulated cysteine, necessary for protein dephosphorylation. ROS-induced oxidation of this cysteine residue renders PTP totally inactive [Tonks, 2005]. In particular, studies have shown ability of hydrogen peroxide, superoxide and peroxynitrites to inhibit PTP rapidly and irreversibly [Lee and Esselman, 2002; Paravicini and Touyz, 2006]. Since a role of PTP inhibition has recently been shown in PH, in particular in signalling pathways activated by growth factors such as PDGF [ten Freyhaus et al. 2011], redox-dependent inhibition of PTP by ROS might contribute to these mechanisms.
Serine/threonine kinases
Mitogen-activated protein kinases (MAPK) are a family of serine/threonine kinases that control many cellular effects such a proliferation, survival, differentiation, apoptosis and stress signals [Griendling et al. 2000]. Activation of MAPK depends on many upstream phosphorylations, leading to activation of multiple signalling pathways. MAPK kinase redox sensitivity has been known for a long time, since exogenous ROS such as hydrogen peroxide and superoxide are able to activate MAPK in endothelial [Huot et al. 1997, 1998] and smooth muscle cells [Ushio-Fukai et al. 1998; Yoshizumi et al. 2000]. In the vasculature, all of the major MAPK (extracellular-regulated protein kinase or ERK, p38 and c-jun N-terminal kinase [JNK]) are activated by growth factors such as angiotensin II or PDGF [Sundaresan et al. 1995; Ushio-Fukai et al. 1998]. More recently, studies have shown that intracellular ROS, mainly derived from NADPH oxidases, are involved in these signalling pathways [Meloche et al. 2000; Torrecillas et al. 2001; Viedt et al. 2000]. Less is known about redox-induced regulation of MAPK in the pulmonary vasculature. However, ROS have been shown to activate ERK and p38 pathways in PAEC [Usatyuk et al. 2003]. Inversely, ROS synthesis in PAEC by NADPH oxidases is dependent upon activation of ERK and p38 pathways [Parinandi et al. 2003]. These results therefore suggest an interrelation between ROS and MAPK in pulmonary endothelial cells after activation by growth factors.
Rho kinases (ROCK) are serine/threonine kinases involved in the regulation of vascular tone, and in the development of several cardiovascular diseases [Perez-Vizcaino et al. 2010]. Two isoforms have been identified and named ROCK-1 and ROCK-2, and both are expressed in the vascular smooth muscle [Loirand et al. 2006]. ROCKs are activated by the small G protein RhoA and then phosphorylate the myosin light chain phosphatase (MLCP), which becomes inactive. Myosin phosphorylation is therefore increased, thus increasing contraction of the smooth muscle, independently of any increase in cytosolic calcium concentrations, a mechanism termed calcium sensitization [Resta et al. 2010]. Such calcium sensitization through activation of the RhoA/ROCK pathway may contribute to the smooth muscle dysfunction observed in PH [Connolly and Aaronson, 2011; Oka et al. 2008], and a relationship between ROS and the RhoA/ROCK pathway has been described in pulmonary arteries [Connolly and Aaronson, 2011; Oka et al. 2008]. Indeed, in chronic hypoxia, RhoA activity is enhanced and is related to increased ROS production [Broughton et al. 2010; Jernigan et al. 2008]. Moreover, a superoxide generator causes ROCK-2 translocation to the nucleus in rat pulmonary arteries and induces a vasoconstriction sensitive to a ROCK inhibitor [Knock et al. 2009]. Hydrogen peroxide may also be able to increase RhoA activity in PAECs and PASMCs [Chi et al. 2010] although controversial results have been observed in other studies [Knock and Ward, 2011; Perez-Vizcaino et al. 2010].
The protein kinase C family contains several isoforms belonging either to conventional PKC or cPKC (α, β1, β2 and γ), to novel PKC or nPKC (δ, ϵ, η, θ, υ, and µ) or to atypical PKC or aPKC (ζ and λ/ι) [Reyland, 2009]. In pulmonary arteries, various PKC isoforms are expressed in fibroblasts, endothelial and smooth muscle cells [Dempsey et al. 2007]. Hydrogen peroxide and peroxynitrites can activate PKC in PASMCs [Chakraborti and Michael, 1993]. In addition, hydrogen-peroxide-induced calcium sensitization is associated with PKCα activation, and is blocked by PKC inhibitors [Pourmahram et al. 2008]. Moreover, hypoxia-induced ROS production is PKC-dependent in PASMCs [Rathore et al. 2008] and in PAECs [Chatterjee et al. 2011]. These results therefore suggest a role of ROS in PKC-dependent signalling pathways in pulmonary vascular cells.
The serine/threonine kinase Akt, also called protein kinase B or PKB, is a downstream target of phosphatidylinositol-3 kinase, and plays a major role in various cellular processes, including cell proliferation and survival, as well as in protein synthesis [Coffer et al. 1998]. A relation between Akt and ROS has been evidenced in systemic vascular smooth muscle cells from various origins, since exogenous hydrogen peroxide induces Akt activation in these cells [Ushio-Fukai et al. 1999]. In addition, angiotensin-II-induced activation of Akt is inhibited by catalase, suggesting a role for ROS in agonist-induced Akt activation [Ushio-Fukai et al. 1999]. In the pulmonary vasculature, ROS generation by NADPH oxidase in PASMC leads to redox-sensitive activation of Akt [Djordjevic et al. 2005]. Moreover, in the same cells, expression of the fibroblast growth factor-2 (FGF-2) is mediated by an increase in superoxide levels via NADPH oxidase activation, a mechanism that is blocked by a pharmacological inhibitor of Akt [Black et al. 2008]. In addition, chronic intermittent hypoxia causes PH and is associated with increased lung levels of NADPH oxidases as well as increased activity of PDGFR and Akt [Nisbet et al. 2009]. These mechanisms are reduced in Nox2 knockout mice [Nisbet et al. 2009], therefore suggesting a role for NADPH oxidase-derived ROS in Akt activation associated with PH.
Other candidate enzymes
Many other enzymes appear to be redox sensitive and are activated by addition of exogenous ROS [Griendling et al. 2000]. For example, phospholipase D is activated by hydrogen peroxide in endothelial cells[Natarajan et al. 1993], and PDGF-induced activation of STAT 1 is inhibited by antioxidants [Simon et al. 1998]. In addition, activation of many other kinases such as Fyn kinase, Proline-rich tyrosine kinase (Pyk) or JAK, seem to be ROS-dependent in vascular tissues from other origins than the lung and may therefore also contribute to the regulation of pulmonary vascular tone in physiological and pathophysiological conditions [Knock and Ward, 2011].
Ion channels
Voltage-gated potassium channels (Kv) play an important role in the regulation of pulmonary vascular tone, and their inhibition seems to be involved in PH pathophysiological mechanisms [Moudgil et al. 2006]. Effects of ROS on Kv channels have been evidenced in pulmonary arteries. Indeed, in PASMC, hypoxia induces ROS production and leads to inhibition of Kv channels [Bonnet and Archer, 2007]. Moreover, hydrogen peroxide homologues also block Kv channel activity, whereas hypoxia-induced Kv channel inhibition is prevented by catalase [Cogolludo et al. 2006].
Transient receptor potential (TRP) channels are composed of several subfamilies and contribute to cation entry in pulmonary arteries [Dietrich and Gudermann, 2011; Guibert et al. 2011]. In particular, expression of TRP channel subtypes belonging to the canonical (TRPC), melastatin-related (TRPM) and vanilloid-related (TRPV) subfamilies have been identified in pulmonary arteries [Yang et al. 2010]. Although direct evidence of TRP modulation by ROS in pulmonary arteries is lacking, ROS-induced regulation of TRP channels has been observed in other vascular tissues [Song et al. 2011], and may therefore contribute to regulation of calcium entry, in particular in PASMC.
Redox-sensitive gene expression
Many cardiovascular genes are redox sensitive, with ROS regulating expression of adhesion molecules, chemotactic factors, antioxidant enzymes and vasoactive substances [Griendling et al. 2000]. This regulation occurs at various levels. Redox sensitivity of some genes can lead to activation of their transcription by ROS-dependent signalling pathways [Wung et al. 1999]. ROS can also modify the activity of specific transcription factors, modulating their turnover, expression, translocation or affinity to their cognate DNA-binding sites. This has been shown in systemic vascular cells in particular for the nuclear factor (NF)-κB [Morgan and Liu, 2011] and the activator protein-1 (AP-1) [Barchowsky et al. 1995; Remacle et al. 1995], but also for the hypoxia-inducible factor (HIF)-1, known to play an important role in hypoxia-induced PH [Huang et al. 1996].
Most of the redox-sensitive genes identified so far are responsive to externally applied oxidant stress, but some of them have also been demonstrated to be downstream of an endogenous source of ROS, such as genes for angiotensin II, PDGF or tumor necrosis factor-α (TNF-α) [Griendling et al. 2000], or genes for many other inflammatory cytokines and adhesion molecules [Paravicini and Touyz, 2006]. Since all of these molecules play an important role in PH development and persistence, these data suggest that regulation of gene expression by oxidative stress may be important in pulmonary vascular cells.
Important role of ROS in the pathogenesis of PH
Sources of ROS in PH
Oxidative stress in the pulmonary vasculature has been evidenced in many PH animal models. In chronic hypoxia-induced PH, an increase in ROS production has been shown in pulmonary arteries [Fresquet et al. 2006; Hoshikawa et al. 2001; Jankov et al. 2008; Liu et al. 2006]. In addition, many of the pathological changes in the pulmonary vasculature observed in this model can be reversed by the administration of antioxidants [Elmedal et al. 2004; Hoshikawa et al. 2001; Lai et al. 1998]. In the rat model of PH induced by monocrotaline (an alkaloid extracted from the plant Crotalaria spectabilis that, once metabolized in the liver, induces pulmonary endothelial cell necrosis and leads to severe PH), elevated levels of superoxide have also been reported in pulmonary arteries [Kamezaki et al. 2008; Mathew et al. 2002]. In this model as well, antioxidant therapies attenuate PH development and decrease right ventricular hypertrophy [Kamezaki et al. 2008; Redout et al. 2010]. In addition, elevated superoxide levels have been reported in other PH models [Brennan et al. 2003; Grobe et al. 2006]. These results are in accordance with data observed in humans where increased ROS levels associated with decreased activity of antioxidant enzymes such as Mn SOD have been reported in the lungs of PH patients [Bowers et al. 2004; Demarco et al. 2010; Spiekermann et al. 2009]. These data therefore suggest that chronic oxidative stress is present in lungs of PH patients.
In accordance with increased ROS levels detected in the pulmonary vasculature during PH, expression and/or activity of various sources of ROS is increased in pulmonary arteries. Several studies have reported an important role of NADPH oxidases, and in particular of several Nox subunits, in various PH animal models [Dennis et al. 2009; Grobe et al. 2006; Redout et al. 2007]. Indeed, Nox2 knockout mice fail to develop hypoxia-induced PH [Liu et al. 2006], and these mice were in particular protected against the development of endothelial dysfunction in intrapulmonary arteries [Fresquet et al. 2006]. Hypoxia-induced PH in mice was also associated with increased lung levels of Nox4 [Mittal et al. 2007; Nisbet et al. 2009]. In lungs from patients with idiopathic pulmonary arterial hypertension, expression levels of Nox4 were upregulated [Mittal et al. 2007]. In vitro, a significant increase of Nox4 expression has been observed under hypoxic conditions in human pulmonary artery adventitial fibroblasts [Li et al. 2008] and in PASMC [Mittal et al. 2007]. Nox4 expression was further increased under hypoxic conditions in pulmonary artery adventitial fibroblasts isolated from the lungs of patients with idiopathic pulmonary arterial hypertension compared with control donors [Li et al. 2008]. Altogether, these results therefore suggest a preponderant role for NAPDH oxidase containing Nox2 and/or Nox4 in oxidative stress in PH.
Some data also provide evidence of eNOS uncoupling in pulmonary arteries, contributing to the generation of elevated levels of ROS during PH. Data obtained in mutant models underline that local BH4 availability is crucial in maintaining pulmonary vascular homeostasis and plays a pivotal role in the pathogenesis of PH [Khoo et al. 2005; Nandi et al. 2005]. Indeed, increased BH4 synthesis prevents PH development. Conversely, BH4 deficiency elevates pulmonary vascular tone by decreasing eNOS activity and NO bioactivity, and promotes pulmonary vascular remodelling, PH and right ventricular hypertrophy [Khoo et al. 2005; Nandi et al. 2005]. These data were obtained in genetically modified mice models, in which tissue BH4 levels are low because of reduced expression of GTPCH-1, or in which endothelial BH4 synthesis is augmented by targeted overexpression of GTPCH-1. However, only few studies have investigated whether alterations of biopterin metabolism and eNOS uncoupling occur in pathophysiological relevant PH models: in lamb models of persistent PH of the newborn, increases in oxidative stress due to uncoupled eNOS and elevated levels of BH2 likely contribute to impaired NO-dependent pulmonary vasodilation [Grobe et al. 2006; Konduri et al. 2007]. Other studies also provide evidence of eNOS uncoupling in pulmonary arteries, contributing to generate elevated levels of ROS during PH [Gielis et al. 2011; Jerkic et al. 2011; Konduri et al. 2007; Lakshminrusimha et al. 2007; Toporsian et al. 2010; Wunderlich et al. 2008].
Altered expression and/or activity of other ROS sources in pulmonary arteries have also been reported. For example, activity of XO is increased, leading to elevated ROS levels in vitro in cultured pulmonary vascular cells [Terada et al. 1992], and in vivo as well with increased nitrotyrosine formation in pulmonary arteries [Jankov et al. 2008].
ROS and altered pulmonary vascular reactivity
Many studies have shown evidence of a role of ROS in altered pulmonary vascular reactivity observed in PH (Figure 4). Indeed, both increased reactivity to contractile agents and endothelial dysfunction can be prevented or reversed by antioxidant treatments [Elmedal et al. 2004; Lai et al. 1998; Liu et al. 2006]. An important role for NADPH oxidase-derived ROS has been evidenced in some studies. Indeed, hypoxia-induced amplification of the responses of pulmonary arteries to various vasoconstrictors is decreased in Nox2 knockout mice [Liu et al. 2006]. As demonstrated by our group, these Nox2 knockout mice are also protected against development of endothelial dysfunction in intrapulmonary arteries [Fresquet et al. 2006]. Therefore, these data show a role for Nox2-derived ROS in exacerbated vasoconstriction and endothelial dysfunction induced by chronic hypoxia in pulmonary arteries. Nox4 also plays a role in hypoxia-induced contraction of mice [Rathore et al. 2008] or of bovine pulmonary arteries [Ahmad et al. 2010]. Indeed, Nox4 is activated by hypoxia and increases intracellular ROS and calcium levels to participate to PASMC contraction [Rathore et al. 2008].
Data obtained in our group reveal an endothelial dysfunction occurring within the intrapulmonary arteries of hypoxic mice, despite marked upregulation of eNOS expression (Figure 5). Moreover, these data also show that, even though hypoxic Nox2 knockout mice did not develop endothelial dysfunction, endothelium-dependent relaxation to acetylcholine was not improved in hypoxic wild-type mice in the presence of ROS scavengers such as cell-permeant SOD or catalase [Fresquet et al. 2006] (Figure 5). These results therefore suggest that impairment of endothelial NO-dependent relaxation in pulmonary arteries from hypoxic mice is not due to inactivation of NO by ROS. This is further supported by the finding that β2-adrenergic receptor-induced relaxation is not altered in intrapulmonary arteries from hypoxic mice, even if this relaxation, similarly to the one induced by acetylcholine, is mediated by endothelial NO [Leblais et al. 2008] (Figure 5). Our data even show that relaxation induced through activation of β2-adrenergic receptors or through the exogenous NO donor sodium nitroprusside may rather be increased in intrapulmonary arteries from hypoxic mice [Leblais et al. 2008] (Figure 5). Altogether, these data indicate that NADPH-oxidase derived ROS may act as a trigger rather than a direct mediator of endothelial dysfunction in intrapulmonary arteries during hypoxic PH. Moreover, since activation of pulmonary β2-adrenergic and muscarinic receptors both lead to eNOS activation, but through activation of different signalling pathways [Banquet et al. 2011; Leblais et al. 2008], mechanisms underlying eNOS activation might therefore be key determinants of pulmonary endothelial dysfunction occurring in PH.
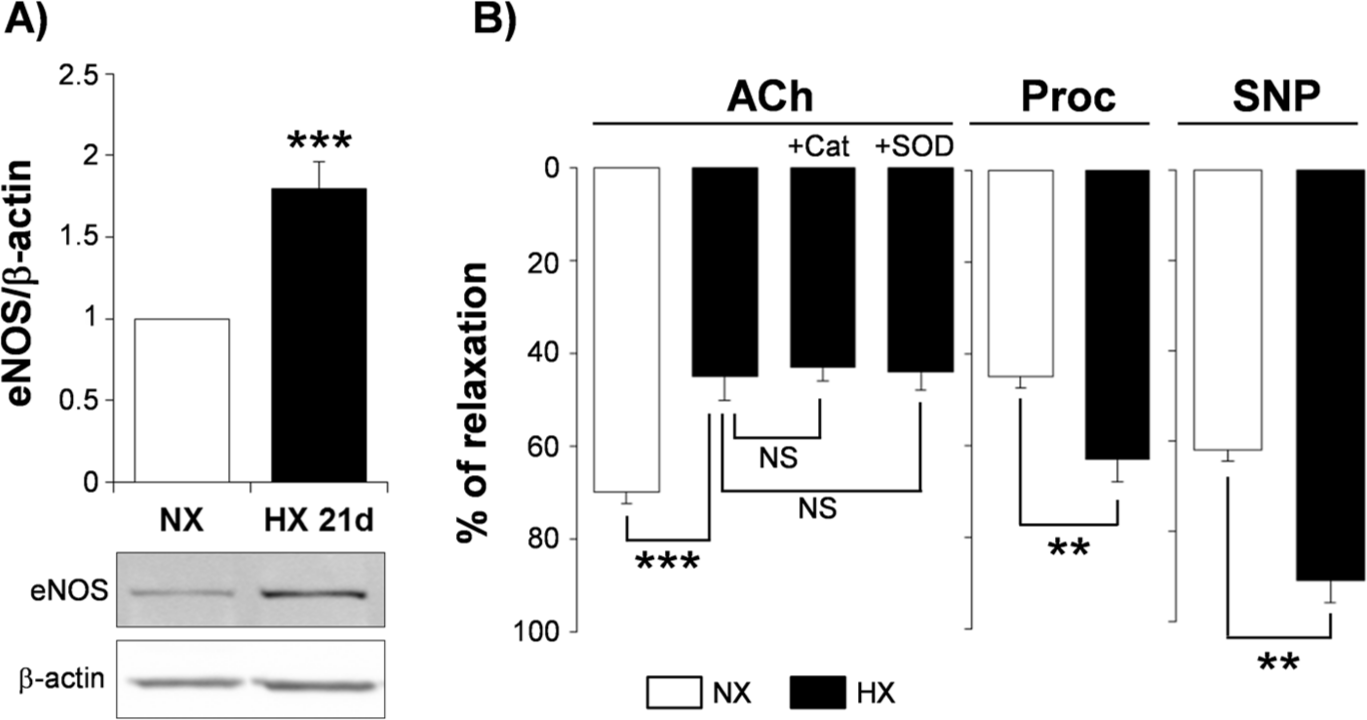
ROS are triggers rather than direct mediators of endothelial dysfunction in hypoxic pulmonary hypertension.
In addition to endothelial dysfunction, pulmonary arteries often exhibit hyperreactivity to contractile agents in PH. Isoprostanes (products from ROS-mediated peroxidation of fatty acids) may participate in these vasomotor alterations. Indeed, as recently demonstrated by our group, release of isoprostanes by pulmonary arteries is increased during chronic hypoxia [Delannoy et al. 2010]. Moreover, pulmonary arterial hyperreactivity to phenylephrine in hypoxic mice is decreased by catalase and by TP receptor antagonists. These results therefore show that ROS contribute to pulmonary arterial hyperreactivity to contractile agents in PH via isoprostane production and further activation of TP receptors [Delannoy et al. 2010]. Such mechanism has also been recently observed in newborn piglets with chronic hypoxia-induced PH [Fike et al. 2011].
ROS and pulmonary vascular remodelling
Studies have evidenced a role of ROS in pulmonary vascular remodelling observed in PH, with in particular an important role of NADPH oxidase-derived ROS (Figure 4). Indeed, Nox2 knockout mice are protected against development of vascular remodelling in chronic hypoxia-induced PH [Liu et al. 2006], and Nox4 participates in hypoxia-induced pulmonary vascular remodelling [Mittal et al. 2007]. ROS generation from other sources may also contribute to pulmonary vascular remodelling. For example, the increased pulmonary vascular remodelling in a mouse model of familial PH is associated with eNOS-dependent ROS production [Jerkic et al. 2011].
Mechanisms activated by ROS and participating in pulmonary vascular remodelling are various and involve all pulmonary vascular cell types [Frazziano et al. 2012]. Medial thickening observed in pulmonary arterial remodelling is characterized in particular by increased proliferation and decreased apoptosis of PASMCs [Hassoun et al. 2009]. PASMC proliferation can be directly stimulated by ROS such as superoxide and hydrogen peroxide [Wedgwood et al. 2001]. Conversely, antioxidants such as superoxide dismutase or catalase can inhibit proliferation of PASMC, and even induce their apoptosis in the long term [Wedgwood and Black, 2003, 2004; Wedgwood et al. 2001]. In addition, ROS contribute to PASMC proliferation triggered by hypoxia, by directly involving activation of Nox4 [Mittal et al. 2007]. Hypoxia-induced ROS generation in PASMC also contributes to increased expression and activity of the hypoxia-inducible factors (HIF)-1αand HIF-2α, involved in PASMC proliferation [Diebold et al. 2010a, 2010c]. Interestingly, activation of HIF-1α by hypoxia in PASMC increases ROS production in these cells through activation of Nox4 [Diebold et al. 2010c] and Rac-1 transcription [Diebold et al. 2010b; Patil et al. 2004]. These data therefore suggest the existence of a positive feedback loop that may contribute to increase PASMC proliferation in hypoxic conditions. This positive feedback loop has also been demonstrated for HIF-2α in PASMC [Diebold et al. 2010a]. Finally, ROS-induced activation of HIF-1α in PASMC also increases expression of several factors, such as FGF-2, that can exert an autocrine proliferative effect and contribute to increased PASMC proliferation in hypoxic conditions [Black et al. 2008]. Moreover, hypoxia-induced PASMC proliferation also involves increased mitochondrial ROS production with opening of KATP channels and mitochondrial depolarization [Hu et al. 2010]. Finally, ROS can activate other transcription factors, such as for example the redox-sensitive transcription factor early growth response-1 (Egr-1), involved in hypoxia-induced PASMC proliferation [Hartney et al. 2011]. ROS also contribute to PASMC proliferation induced by several vasoactive factors. For example, endothelin-1 stimulates PASMC proliferation via NADPH oxidase-catalysed ROS production [Wedgwood et al. 2001]. Activation of NADPH oxidase by serotonin stimulates the proliferation of bovine PASMC via a pathway involving the MAP kinases ERK1/2 [Lee et al. 1999, 2001]. Several growth factors have also been shown to activate NADPH oxidase in vascular smooth muscle cells, including angiotensin II [Griendling et al. 1994] and PDGF [Marumo et al. 1997]. In PASMCs, the transforming growth factor-β1 (TGF-β1) activates Nox4 and increases ROS production that contributes to TGF-β1-induced cell proliferation [Ismail et al. 2009; Sturrock et al. 2006]. TGF-β1-induced ROS production in PASMC also contributes to upregulated expression of other growth factors, such as the vascular endothelium growth factor (VEGF) [Mata-Greenwood et al. 2005]. These results therefore suggest that ROS-mediated signalling pathways contribute to increase PASMC proliferation stimulated by various contractile and growth factors whose expression is increased in PH. As described recently, iron may play an important role in these ROS-dependent signalling pathways for PASMC growth [Wong et al. 2012].
ROS may also be involved in pulmonary vascular remodelling through participating to the excessive proliferation of endothelial cells. Indeed, ROS directly stimulates PAEC proliferation [Browning et al. 2012; Milovanova et al. 2004, 2006], and triggers angiogenesis [Nijmeh et al. 2010]. Conversely, antioxidants can inhibit PAEC proliferation [Han et al. 2010; Milovanova et al. 2006]. ROS may also contribute to PAEC survival induced by other factors, such as the endogenous cytochrome P-450 product 20-hydroxyeicosatetraenoic acid (20-HETE) [Dhanasekaran et al. 2009; Medhora et al. 2008]. Finally, oxidative stress can also activate HIF-1α in PAEC and therefore increase expression of factors such as PDGF that participate in pulmonary vascular remodelling [Mermis et al. 2011].
ROS have been shown to participate to many signalling pathways in adventitial fibroblasts from various origins [Haurani and Pagano, 2007]. The role of the adventitial layer in vascular inflammation and remodelling is now largely studied, in particular in PH [Stenmark et al. 2012]. ROS stimulate proliferation and inhibit apoptosis of pulmonary artery adventitial fibroblasts [Li et al. 2008], and may therefore contribute to adventitial remodelling observed in PH [Stenmark et al. 2012].
Finally, modulation of the extracellular matrix by matrix metalloproteinases (MMPs) is also an important component in vascular remodelling of the pulmonary vasculature [Chelladurai et al. 2012]. MMPs are a family of enzymes capable of degrading components of the extracellular matrix, and their activity is tightly regulated at transcriptional and protein synthesis levels. MMP-2 and MMP-9 expression and activity are increased in PH [Cantini-Salignac et al. 2006; Frisdal et al. 2001; Lepetit et al. 2005; Umar et al. 2007], and MMP-9 overexpression in transgenic mice augments PH [George and D’Armiento, 2011]. NAPDH oxidase-dependent generation of ROS in PAEC leads to MMP activation [Cook-Mills, 2006]. Moreover, a role for ROS has recently been established in right ventricular failure observed in PH, through induction of MMP activity [Qipshidze et al. 2011]. These results therefore suggest that redox-sensitive mechanisms occur in the pulmonary vasculature for MMPs, and may contribute to pulmonary arterial remodelling.
ROS and pulmonary vascular inflammation
Inflammation is a common feature in remodelling vessels in PH, characterized by an accumulation of perivascular inflammatory cells, including macrophages, dendritic cells, T and B lymphocytes, and mast cells, together with elevated circulating levels of various cytokines and chemokines [Price et al. 2012]. In the lung, ROS activate redox-sensitive transcription factors such as AP-1, HIF-1 of NF-κB to initiate inflammatory responses [Lee and Yang, 2012; Park et al. 2009]. For example, in response to oxidative stress, lung cells release pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, IL-8 and TNF-α [Rahman and MacNee, 2000]. Moreover, expression of many inflammatory target proteins triggered by pro-inflammatory cytokines such as IL-1β or TNF-α occurs through increased ROS production [Lee et al. 2011; Phalitakul et al. 2011]. Increased ROS levels in the lung also augment vascular leak by stimulating endothelial permeability, thus exacerbating inflammation [Pearse et al. 2003; Usatyuk et al. 2003]. ROS also participate in hypoxia-induced pulmonary vascular leak [Wojciak-Stothard et al. 2005]. This effect involves increased expression and activation of the transient receptor potential melastatin (TRPM)2 [Frey et al. 2009; Hecquet et al. 2008], as well as increased VEGF expression, triggered by ROS-induced HIF-1α activation [Irwin et al. 2009; Lee et al. 2006]. Finally, a direct role of pro-inflammatory cytokines such as IL-1β or TNF-α has been recently shown in endothelial dysfunction, involving activation of NADPH oxidase and ROS production in endothelial cells [Barbieri et al. 2011]. These results therefore suggest that oxidative stress, by regulating inflammatory signalling pathways and expression of inflammatory targets in the lung, may participate in vascular inflammation in PH (Figure 4).
Future directions and conclusions
Over recent years, a large body of evidence has shown an increased oxidative stress in PH, with ROS playing multiple roles in the pathogenesis of this disease. Current PH treatments, mainly designed to target altered pulmonary arterial reactivity, manage to slow the progression of the disease but do not afford a cure. In this context, targeting ROS seems to be an interesting therapeutic approach. Indeed, ROS participate in pulmonary vascular altered reactivity, but are also involved in pulmonary vascular inflammation and remodelling, two other major PH features. Several therapeutic strategies have therefore been developed to reduce ROS levels in PH [Frazziano et al. 2012; Tabima et al. 2012].
A first strategy developed is to use ROS scavengers. For example, catalase and SOD mimetics decrease ROS levels and exert preventive effects in experimental PH [Archer et al. 2010; Delannoy et al. 2010]. In addition, use of recombinant human SOD [Farrow et al. 2008] and gene transfer of extracellular SOD [Kamezaki et al. 2008; Nozik-Grayck et al. 2008] have shown beneficial effects in PH animal models.
Another strategy is the disruption of sources of ROS in the pulmonary vasculature. Use of several pharmacological inhibitors have shown beneficial effects in experimental PH, such as inhibition of uncoupled eNOS with L-NAME [Wunderlich et al. 2008], or inhibition of XO with allopurinol [Jankov et al. 2008]. Therapies targeting the altered mitochondrial metabolism are also investigated and constitute a promising approach, as shown by dichloroacetate, an inhibitor of mitochondrial pyruvate dehydrogenase kinase that displays curative effects in PH animal models [Bonnet et al. 2006; McMurtry et al. 2004]. Another approach consists of targeting the fatty acid oxidation pathway, through activation of the peroxisome proliferator-activated receptor (PPAR)γ [Rabinovitch, 2010]. The PPARγ agonist rosiglitazone attenuates hypoxia-induced PH in the rat [Kim et al. 2010], through regulation of NF-κB and HIF-1α activity [Kang et al. 2011], and leading in particular to decreased Nox4 expression and decreased ROS production [Lu et al. 2010]. These results therefore suggest PPARγ as a potential PH therapeutic target. However, adverse cardiovascular and cancer events detected in diabetic patients for rosiglitazone and pioglitazone, respectively, emphasize the need for caution before employing these agents in PH patients [Green et al. 2011].
Finally, particular attention is given to NADPH oxidases. Indeed, Nox enzymes are involved in many pulmonary vascular mechanisms, and recent findings suggest their pivotal role in PH. NADPH oxidase inhibitors prevent and reverse PH in animal models [Dennis et al. 2009; Liu et al. 2006]. Further evidence comes from genetically modified mice lacking NAPDH oxidase and therefore protected from experimental PH [Fresquet et al. 2006; Lee et al. 2006]. Development of specific Nox inhibitors designed to modulate Nox isoforms in pulmonary vascular cells may therefore be of interest in PH patients [Frazziano et al. 2012].
In conclusion, many studies have shown enhanced oxidative stress associated with PH, and evidenced an important role of ROS in various pathophysiological aspects of this disease. New therapeutic approaches in PH targeting ROS through blockade of ROS-dependent signalling pathways, or through disruption of sources of ROS in the pulmonary vasculature, targeting in particular Nox enzymes, remain therefore promising.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.