
Abstract
Cardiovascular diseases (CVDs) remain the foremost global cause of mortality, largely driven by endothelial dysfunction and vascular remodelling. The RhoA/Rho-associated coiled-coil containing protein kinase (ROCK) signalling pathway is central to vascular homeostasis, regulating vascular smooth muscle cell (VSMC) phenotypic plasticity, contraction, nitric oxide (NO) bioavailability, and cytoskeletal organization. Aberrant RhoA/ROCK activation contributes to hypertension, atherosclerosis, heart failure, and vascular stiffness through impaired endothelial function, oxidative stress, and maladaptive remodelling. This narrative review summarises current evidence on molecular mechanisms linking RhoA/ROCK activity with endothelial dysfunction and vascular pathology, highlighting its crosstalk with phosphoinositide-3-kinase–protein kinase B/Akt (PI3K-PKB/Akt), reactive oxygen species (ROS), and inflammatory mediators. Therapeutic insights focus on ROCK inhibitors, particularly fasudil, their limitations in isoform selectivity, and recent advances in structure-based drug design and targeted delivery systems. Isoform-specific and next-generation inhibitors represent a new frontier in therapy, with the potential to precisely suppress pathogenic RhoA/ROCK-driven inflammation, restore endothelial function, and markedly diminish cardiovascular disease burden.

Keywords
1. Introduction
Vascular smooth muscle cell (VSMC) activity must be properly maintained for the vasculature to operate physiologically and to regulate blood flow and vasotone. VSMCs are quiescent, immobile, and contractile in healthy vasculature. Furthermore, myosin heavy chain, smooth muscle α-actin (SMα-actin), SM 22α, calponin, myosin light chain (MLC), and smoothelin are among the contractile proteins expressed by VSMCs, which are also thought to be differentiated.1,2 Even with their distinct phenotype, VSMCs exhibit plasticity. To preserve the vasculature’s homeostasis, VSMCs may change from the contractile phenotype in response to stimuli. Crucially, for the creation and maturation of arteries, VSMC phenotypic switching is required during vascular development and remodeling. 3
Though mostly physiological, this phenotypic shift may play a role in the processes leading to vascular disease when it is dysregulated. For example, VSMC defective phenotypic alterations are involved in intimal hyperplasia, a process that underlies the pathophysiology of several illnesses, including hypertension, aortic aneurysm, and atherosclerosis. Notably, one of the early signs of vascular disorders is frequently the process of VSMC phenotypic flipping.4–6 Studies using small conditional RNA (scRNA) is helpful in investigating the transcriptome of phenotypically transformed VSMCs and the mechanisms that lead to this alteration. 7 When VSMCs undergo phenotypic switching or a sick vascular state, they dedifferentiate, show aberrant gene expression patterns, and take on a synthetic and proliferative character.8,9 Furthermore, scRNA investigations show that they become more migratory, particularly to sites of cellular damage, prone to clustering and apoptosis. In this context, single-cell RNA sequencing discloses that foam cells produced from VSMC exhibit a tendency to experience many types of cell death, like necroptosis, pyroptosis, autophagy, and apoptosis. These processes collectively contribute to the physio-pathology and anatomical features of atherosclerotic plaques. 9
Since VSMC phenotypic flipping plays a role in the early phases of VD, it makes sense to identify the molecular mechanisms governing this process (Figure 1). Gaining further insight into the molecular mechanisms behind this phenotypic shift might lead to the identification of novel therapeutic targets for treating CVD. Within the framework of VSMC phenotypic switching, this study examines the roles played by the small GTPase RhoA and its effector kinase, Rho-associated kinase (ROCK). Along with outlining the pathway’s current state and demonstrating how it contributes to vascular dysfunction, this narrative review also offers some conjecture on the potential therapeutic advantages of rho-kinase inhibitors. We’ll also review the function of RhoA/Rho-kinase in the vasculature, and its expression and isoforms. This review is guided by the Scale for the Assessment of Narrative Review Articles (SANRA).
10
RhoA/ROCK signaling triggered by vasoactive agonists; endothelin-1 (ET-1), serotonin (5-HT), and norepinephrine (NE) activate G-protein–coupled receptors, leading to G-protein stimulation and the conversion of RhoA-GDP to active RhoA-GTP through GEFs, while GAPs reverse this process. Activated RhoA stimulates Rho-kinase (ROCK), which phosphorylates key downstream targets—including myosin light chain (MLC), ERM proteins, adducin, and LIM kinases—to promote smooth-muscle contraction, cytoskeletal stabilization, and proliferative signaling. ROCK also inhibits myosin light-chain phosphatase (MLCP), sustaining MLC phosphorylation, and modulates CRMP2, adenylate cyclase, and endothelial nitric-oxide synthase (eNOS), integrating cytoskeletal control with nitric-oxide–dependent vascular responses. Overall, the pathway coordinates vascular tone, cytoskeletal remodeling, and growth-associated responses. Created with BioRender.com.
2. RhoA and ROCK role in biological function
The catalytic kinase domain located at the N-terminus of the threonine/serine protein kinase is Rho-kinase. The downstream effector of RhoA is responsible for mediating calcium (Ca+2) sensitization. 11 RhoA small GTPase is known to be involved in a range of physiological cellular processes as the molecular switch for different extracellular signals, like migration, contraction, cell cycle progression, adhesion, and gene expression. One of the best-characterized Rho effectors, Rho-kinase, or ROCK, which comes in two isoforms—ROCK1 (also called p160ROCK or ROKβ) and ROCK 2 (also called ROKα) is how RhoA controls these actions. 12 Not only does RhoA activate Rho-kinase, but arachidonic acid, that is released from SM against various agonists. 13
The ezrin-radixin-moesin (ERM) family, the myosin-binding subunit (MBS) of myosin phosphatase, vimentin (an intermediate filament), adducin, LIM-kinase, and the Na+H+ exchanger, which phosphorylates cofilin, are just a few of the identified targets for Rho-kinase. Myosin light chain phosphatase (MLCP), is one of these primary substrates of Rho-kinase and is physiologically responsible for the dephosphorylation of myosin II (MLC20) light chains. 14 Thus, it is thought that MLCP phosphorylation is a distinguishing feature of Rho-kinase activation. MLC may also be directly phosphorylated by rho-kinase. 15 Overall, Rho-kinase activity is consistent with higher phosphorylation of MLC since MLCP inactivation is linked to increased MLC phosphorylation. Muscle contraction results from the molecular contact between myosin and actin, which is made possible by myosin phosphorylation or activation. 15
Rho-kinase has been demonstrated to be significantly engaged in the contraction of SMC, which happens through 2 primary mechanisms: ‘Ca+2 signaling cascades and RhoA/Rho-kinase signaling routes’. Moreover, RhoA/Rho-kinase can change the contractile system’s Ca+2 sensitivity, and when it is activated, it inhibits endothelial nitric oxide synthase (eNOS), which modifies the generation of nitric oxide (NO). 16 Pathological disorders, primarily vascular illnesses and other pathologies including stroke, hypertension, atherosclerosis, vasospasm, pulmonary hypertension, heart failure, and more recently, cancer, have been linked to the disruption of both processes, according to research conducted on humans and animals.17,18 Rho-kinase signaling is a crucial switch control in the dynamic and fast rearrangement of the actin cytoskeleton, which is a common feature seen in many of these diseases. 19
Although the RhoA/Rho-kinase pathway was extensively studied over the past ten years, many details of this pathway still remain unknown. The suppression of this pathway may have profound therapeutic consequences because RhoA regulates critical physiological processes and has already been linked to the control of inflammation, vascular tone, and oxidative stress. Certain substances have been shown to inhibit Rho-kinase and may be useful as medicines for a number of illnesses. The most commonly utilized in experiments are two non-selective inhibitors, fasudil, also known as Y27632 and H1077. 20 Nonetheless, the precise function of ROCK in the vasculature has up to this point been restricted by the absence of particular pharmacological inhibitors as these inhibitors are unable to differentiate between ROCK isoforms or the diverse processes of ROCK in separate cell components. Nevertheless, by highlighting these systems as potential therapeutic targets, these Rho-kinase inhibitors have made a significant contribution to our understanding of the changed mechanisms in vascular disorders. 21
3. ROCK isoform structure and expression
The molecular mass of serine/threonine kinases, or ROCKs, is 160 kDa. They are expressed in Drosophila, zebrafish, mosquito, Xenopus, bovine, chicken, rat, mouse, C elegans, and human. ROCK-1 and ROCK-2, two isoforms of ROCK encoded by distinct genes, were identified. The genes for ROCK-1 and ROCK-2 are found on chromosomes 2 (2p24) and 18 (18q11.1), respectively. ROCK sequences are made up of a kinase domain that is found at the protein’s amino terminus, a coiled-coil region that contains a pleckstrin-homology (PH) domain with a cysteine-rich domain, and the Rho-binding domain (RBD) (Figure 2). Between ROCK-1 and ROCK-2, there is a 65% amino acid sequence similarity, indicating strong homology. In the kinase domain, the identity exceeds 92%, whereas in the RBD, it is 58%.
12
The ROCKs’ molecular structure. The protein’s amino terminus contains the kinase domain, which is followed by a coiled-coil region that houses a PH and the RBD domain that has a cysteine-rich domain in ROCK sequences. Both ROCK-1 and ROCK-2 have a 65% total amino acid sequence similarity, indicating their strong homology. Created with BioRender.com.
In addition to their overlapping structural features, accumulating evidence indicates that ROCK1 and ROCK2 perform distinct isoform-specific functions in vascular pathology. 22 ROCK2 is increasingly considered the predominant isoform in endothelial dysfunction, where it regulates eNOS phosphorylation, oxidative stress generation, leukocyte adhesion, and inflammatory cytokine signaling. Endothelial-specific inhibition or genetic deletion of ROCK2 has been shown to restore endothelial barrier integrity and suppress vascular inflammation. 23 In contrast, ROCK1 plays a more prominent role in vascular smooth muscle cell (VSMC) proliferation, migration, contraction, and fibrosis, thereby contributing significantly to neointimal hyperplasia and arterial remodeling. Studies using ROCK1-deficient models demonstrate reduced VSMC contractility, extracellular matrix deposition, and fibrotic responses, highlighting its dominant role in vascular structural changes.19,24 These isoform-specific contributions strongly support the therapeutic rationale for developing selective ROCK inhibitors that may provide superior efficacy with fewer off-target effects compared to non-selective agents. 25
Despite the lack of thorough analysis, certain studies have revealed variations in ROCK expression regulation. Angiotensin II (AII) stimulates proteins and ROCKs mRNAs via pathways that are reliant on nuclear factor B (NF-kB), protein kinase C, interleukin-1, and AII type 1 receptor activation. In vivo studies on the coronary arteries of mice receiving constant AII treatment similarly reported upregulation of ROCK. 23 In vitro, nicotine amplifies the stimulatory action of AII on the expression of ROCK-2 mRNA in human coronary VSMC, while estrogen opposes this effect. Thus, it has been proposed that the higher frequency of vascular diseases in smokers and postmenopausal women may be partially explained by changes in ROCK expression in VSMC. 26
Apart from the distribution of ROCK isoforms in different organs and tissues, a number of research have examined the subcellular location of these molecules. ROCKs are mostly found in the cytoplasm, but when RhoA is activated, they partly translocate to the peripheral membrane. 27 Though soluble, ROCKs are mostly present in the cleavage furrow during cytokinesis, at the stress fiber where the PH domain binds to myosin II, at the vimentin intermediate filament network, and at other locations. It is yet unknown what processes lead to ROCKs’ subcellular localization. 28
In order to examine the distribution and function of ROCK isoforms in vivo, mice lacking ROCK-1 and ROCK-2 have recently been produced. 29 Loss of ROCK-1 causes animals to have an omphalocele phenotype and open eyes at birth, whereas loss of ROCK-2 causes placental malfunction, which causes intrauterine growth retardation and foetal mortality. Nonetheless, the mice that survive in both knockout animal groups mature properly and are fertile. With the exception of specific tissues, such the placenta, these data imply that ROCK isoforms act redundantly and raise the prospect that either can functionally compensate for the absence of the other in most systems. Analysis of the cardiovascular phenotype in ROCK isoforms knockout mice is lacking. 29
4. RhoA/Rho-kinase signalling in VSMC contraction
Rho proteins are molecular switches that exist in two distinct states: a GDP-bound inactive state and a GTP-bound active state. Activation of RhoA is initiated downstream of membrane receptors via guanine nucleotide exchange factors (GEFs), which are tightly regulated multidomain proteins responsive to extracellular cues. In contrast, RhoA activity is negatively regulated by GTPase-activating proteins (GAPs), which enhance intrinsic GTP hydrolysis, and guanine nucleotide dissociation inhibitors (GDIs), which sequester Rho proteins away from the membrane. 30
RhoA translocation to the plasma membrane and subsequent activation of downstream effectors contributes primarily to Ca2+ sensitization of vascular smooth muscle cells (VSMCs), thereby promoting contraction without necessarily increasing intracellular Ca2+ levels.
31
One of the principal mechanisms involves inhibition of myosin light chain phosphatase (MLCP), which shifts the balance toward myosin light chain kinase (MLCK)–mediated phosphorylation of MLC. Consequently, vascular contraction or relaxation is determined by the dynamic ratio of MLCK to MLCP activity (Figure 3).32,33 Ca2+-dependent and Ca2+-sensitization pathways in vascular smooth muscle contraction. The schematic depicts GPCR-driven PLC/IP3-mediated Ca2+ release and MLCK activation alongside RhoA/ROCK-dependent Ca2+ sensitization via MLCP inhibition. Integration of these pathways sustains MLC phosphorylation and amplifies vascular smooth muscle contraction. Created with BioRender.com.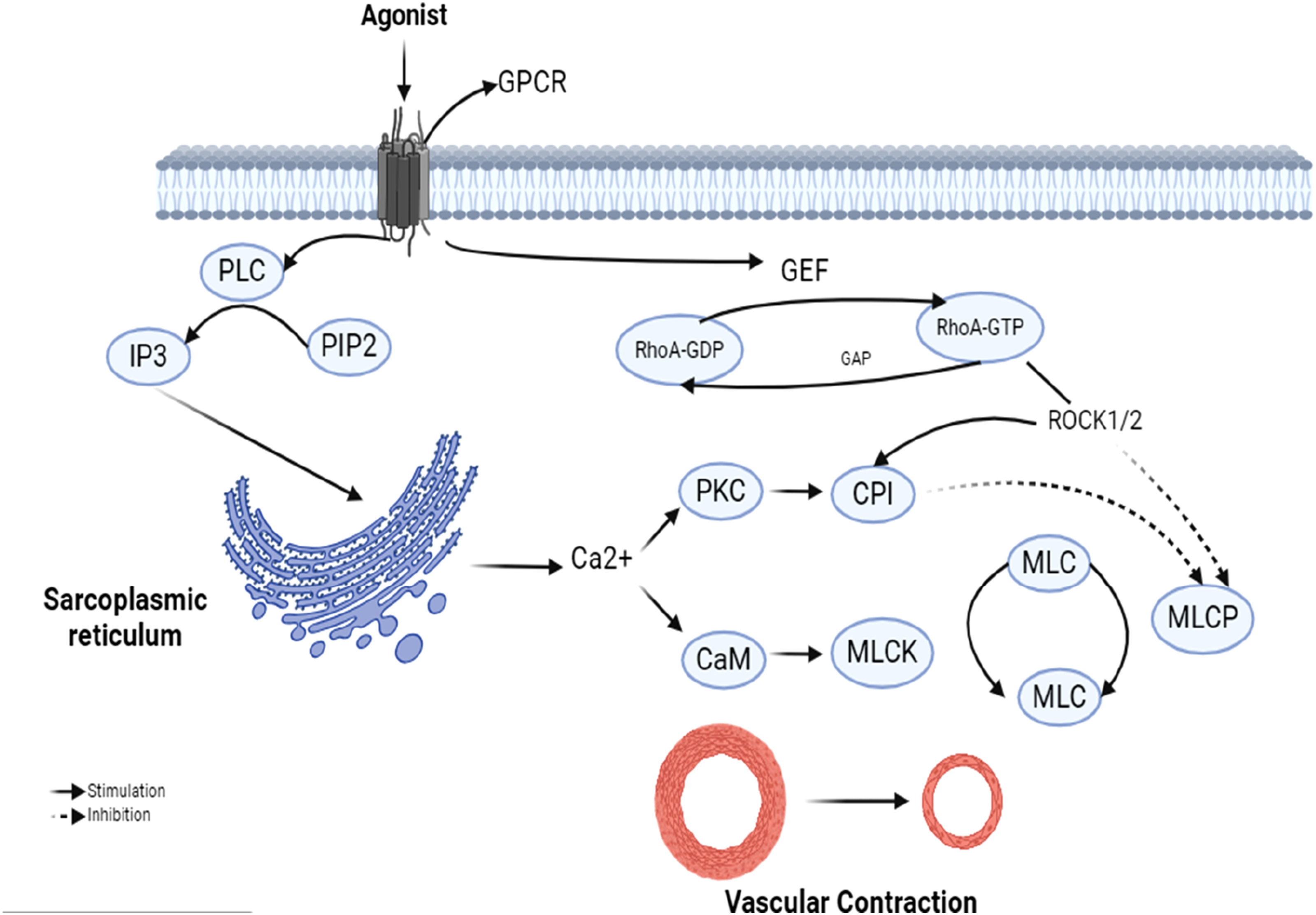
Rho-kinase (ROCK) is the best-characterized downstream effector of RhoA and a central regulator of smooth muscle contraction. 34 Two isoforms, ROCK1 and ROCK2, encoded on chromosomes 18 and 12 respectively, share high homology within their kinase domains but differ in tissue distribution and regulatory interactions. In addition to RhoA, ROCK activity can be modulated by other small G proteins such as Gem, Rad, and RhoE. Notably, RhoE binds directly to the N-terminal catalytic domain of ROCK1, inhibiting its activity and preventing RhoA association. 35
GPCR-driven smooth muscle contraction occurs through two coordinated mechanisms: Ca2+-dependent activation of MLCK and RhoA/ROCK-mediated Ca2+ sensitization. The latter pathway reinforces contractile responses by suppressing MLCP activity and limiting nitric oxide (NO)-mediated relaxation via inhibition of eNOS. Together, these processes ensure efficient and sustained vasoconstriction. 12
ROCK is a serine/threonine kinase of approximately 160 kDa with broad tissue expression, including vascular smooth muscle cells and endothelial cells.36–38 Although both isoforms are expressed in VSMCs, ROCK2 appears to play a dominant role in regulating vascular contractility, whereas ROCK1 contributes to broader developmental and cellular functions.39–41 Genetic knockout studies further support isoform-specific roles, as deletion of ROCK2 results in embryonic lethality, while ROCK1 deficiency leads to placental dysfunction. 37
At the molecular level, ROCK enhances contraction by increasing Ca2+ sensitivity through inhibition of MLCP. MLCP consists of a catalytic subunit (PP1cδ) and the regulatory subunit MYPT1, which governs phosphatase activity toward phosphorylated MLC. 42 ROCK suppresses MLCP either indirectly, by phosphorylating MYPT1 at inhibitory residues (Thr850 and Thr696), or directly by promoting phosphorylation of MLC.43,44 In addition, ROCK and protein kinase C (PKC) can activate CPI-17, a smooth muscle–specific inhibitor of MLCP, further reinforcing contractile signaling. 45
CPI-17–mediated inhibition of MLCP, together with MYPT1 phosphorylation, represents a key determinant of smooth muscle Ca2+ sensitization and sustained contraction. 46 Importantly, the relative contribution of these inhibitory mechanisms varies across vascular beds, highlighting tissue-specific regulation of RhoA/ROCK signaling. Emerging evidence also suggests that ROCK expression and activity can be regulated at both transcriptional and epigenetic levels, as observed in AngII-stimulated VSMCs. 19
Beyond experimental models, clinical studies increasingly demonstrate that VSMC phenotypic transformation is closely linked to subclinical inflammation in hypertension, metabolic syndrome, and atherosclerosis. Biomarker-based inflammatory risk scores, including endothelial activation and stress indices such as EASIX, have been associated with vascular remodeling, atherosclerotic burden, and adverse cardiovascular outcomes. These findings support a clinically relevant link between inflammation-driven VSMC dysfunction and RhoA/ROCK signaling as a contributor to progressive vascular disease. 47
Beyond classical kinase signaling, ROCK activity is modulated by lipid mediators such as arachidonic acid and by redox-dependent post-translational modifications. 48 Under physiological conditions, reactive oxygen species (ROS) act as reversible second messengers; however, in pathological states, excessive ROS amplify ROCK-driven pro-contractile signaling, leading to altered vascular reactivity and enhanced vasoconstriction. 49
Collectively, these findings underscore that RhoA/ROCK signaling primarily augments vascular smooth muscle contraction by enhancing Ca2+ sensitivity rather than by directly elevating intracellular Ca2+ levels. Continued identification of context-specific regulatory mechanisms will further refine therapeutic strategies targeting ROCK-dependent vascular dysfunction (Figure 4). Overview of signaling pathways regulating VSMC contraction and relaxation, highlighting interactions among RhoA/ROCK, Ca2+, MLCK/MLCP balance, and redox-sensitive modulators. Created with BioRender.com.
5. RHOA/ROCK pathway and endothelium-dependent relaxation
5.1. Bioavailability of RhoA/Rho kinase and nitric oxide (NO)
Reduced NO bioavailability is a hallmark of endothelial dysfunction and may arise from impaired eNOS expression, reduced eNOS activation, or oxidative inactivation of NO. Substantial evidence demonstrates that the RhoA/ROCK pathway negatively regulates eNOS expression and activity. For example, activation of RhoA/ROCK in hypertensive profilin-1 transgenic mice markedly suppresses eNOS phosphorylation and NO production. Similarly, thrombin reduces eNOS mRNA stability in human endothelial cells through RhoA/ROCK activation. 50
Pharmacological inhibition of RhoA or ROCK, including treatment with statins, enhances eNOS expression by prolonging eNOS mRNA half-life and restoring NO bioavailability in both experimental models and clinical settings. In ovine fetal pulmonary arteries, the ROCK inhibitor Y-27632 significantly augmented normoxia-induced NO generation. 51 Conversely, geranylgeranyl pyrophosphate reverses statin-induced eNOS upregulation by reactivating RhoA through isoprenylation. 52
5.2. Regulation of signal transduction
Multiple signaling pathways converge on endothelial NO regulation, many of which intersect with RhoA/ROCK signaling. Key contributors include oxidative stress, arginase activity, and the phosphoinositide 3-kinase (PI3K)/Akt pathway, all of which modulate endothelial homeostasis and vascular tone. 52
5.3. RhoA/ROCK pathway and PI3K/Akt
Akt (protein kinase B), a major downstream effector of PI3K, directly phosphorylates eNOS at Ser1177, enhancing NO production. 53 Crosstalk between PI3K/Akt and RhoA/ROCK pathways plays a critical role in endothelial function, independent of ROCK-mediated suppression of eNOS expression. Active RhoA/ROCK signaling inhibits Akt activity, thereby preventing eNOS phosphorylation and reducing NO bioavailability in endothelial cells. 54
Inhibition of RhoA or ROCK rapidly activates PI3K/Akt signaling, leading to increased eNOS phosphorylation and NO release. PTEN, a negative regulator of PI3K, may be directly phosphorylated by ROCK, suggesting a molecular link between RhoA/ROCK signaling and PI3K/Akt modulation.55,56
Beyond the PI3K/Akt axis, AMP-activated protein kinase (AMPK) has emerged as an endogenous counter-regulatory pathway that mitigates RhoA/ROCK-driven endothelial dysfunction. AMPK directly phosphorylates eNOS at Ser1177, enhances NO bioavailability, and suppresses oxidative and inflammatory signaling. 56 Recent evidence indicates that AMPK activation protects against tyrosine kinase inhibitor–induced cardiovascular toxicity by restoring eNOS–NO signaling and redox balance, highlighting its relevance as a compensatory mechanism in vascular homeostasis. 56
5.4. RhoA/ROCK pathway and arginase, ROS
Oxidative stress is a major determinant of reduced NO bioavailability. Superoxide reacts with NO to form peroxynitrite, which oxidizes tetrahydrobiopterin (BH4), uncouples eNOS, and diminishes NO production. Elevated RhoA activity correlates with increased peroxynitrite formation in animal models of vascular disease. 57 ROS also activate RhoA through RhoGEFs, contributing to thrombin-induced endothelial dysfunction and enhanced vascular contraction.58–60
Arginase further limits NO production by competing with eNOS for L-arginine, promoting eNOS uncoupling and superoxide generation.61,62 Experimental and clinical studies demonstrate that activation of the RhoA/ROCK pathway is a prerequisite for increased arginase expression and activity in endothelial cells. Inhibition of RhoA or ROCK suppresses arginase upregulation in hyperglycemic and inflammatory conditions, including TNF-α–stimulated endothelial cells and inflammatory bowel disease.63,64
Clinical evidence further reinforces the central role of oxidative stress in activating the RhoA/ROCK pathway, particularly in metabolic disorders such as diabetes. A recent meta-analysis reports that treatment with rosiglitazone significantly reduces circulating malondialdehyde (MDA), a key biomarker of systemic lipid peroxidation and oxidative burden. 65 This reduction in MDA is clinically relevant because elevated oxidative stress enhances RhoA activation, promotes arginase upregulation, and restricts L-arginine availability for eNOS, collectively amplifying endothelial dysfunction. Thus, the observed MDA-lowering effect of rosiglitazone provides human evidence supporting the concept that reducing systemic oxidative stress can indirectly suppress RhoA/ROCK signaling. 65 Incorporating this translational insight strengthens the argument for targeting redox pathways as a complementary strategy to modulate ROCK-driven vascular injury in diabetes.
Previous research in the lab demonstrated that elevated glucose and diabetes boost arginase activity through improved RhoA/ROCK action. There have also been notable observations of increased RhoA and arginase activity in TNF-α/lipopolysaccharide-activated human EC and inflammatory bowel illness.
64
The suppression of ROCK or RhoA blocks high arginase activity/expression, indicating that activation of the RhoA/ROCK pathway is a necessary prerequisite for elevated arginase expression and activity in the vasculature.
66
Figure 5. Inflammatory and metabolic activation of RhoA/ROCK signaling in VSMCs. This figure presents a proposed signaling cascade whereby inflammatory and metabolic stimuli promote ROS generation and CyPA release, activating PI3K-dependent RhoA/ROCK signaling. These events alter membrane polarization and contractile signaling in VSMCs, contributing to vascular dysfunction under pathological conditions. Created with BioRender.com.
Recent clinical studies further support the relevance of oxidative stress–driven endothelial dysfunction in cardiovascular disease by demonstrating strong associations between redox imbalance and vascular risk. Circulating biomarkers such as malondialdehyde (MDA), total antioxidant capacity (TAC), reduced glutathione (GSH), and superoxide dismutase (SOD) activity are increasingly used to quantify systemic oxidative stress and predict cardiovascular outcomes. Elevated MDA levels and reduced antioxidant defenses correlate with impaired NO bioavailability, endothelial dysfunction, and increased vascular stiffness—hallmarks of RhoA/ROCK pathway activation. Importantly, clinical evidence indicates that redox imbalance not only reflects disease severity but also contributes to coronary microvascular dysfunction and adverse vascular remodeling, highlighting oxidative stress biomarkers as translational indicators of RhoA/ROCK-mediated vascular injury. 67
6. Mechanisms of RhoA/ROCK-induced vascular stiffness
The hardness of the artery wall is connected with vascular stiffness. Since it can result in higher vascular resistance and degradation of the elastic component of artery structure, an increase in vascular stiffness is linked to various CVD. Moreover, end-organ remodeling, like adult arterial intima-media thickening, is linked to elevated arterial stiffness. 68 Changes in the composition of the primary vascular wall constituents—collagen, smooth muscle cells, and elastin—modify vascular stiffness. Arterial stiffness is significantly influenced by the stiffness of VSMCs in and of themselves. 69 In this instance, VSMC and arterial stiffness are both influenced by Rho/ROCK signaling.
Among other signals, the primary cause of VSMC stiffness which appears in hypertension is the transcriptional program that is initiated by the myocardin/SRF transcriptional program. 70 Myocardin nuclear translocation, the activation of the myocardin/SRF transcriptional pathway, and ultimately the transition to a stiffer VSMC phenotype may all be directly induced by Rho/ROCK signaling. This may ultimately result in the blood vessel’s hemodynamic characteristics changing and the entire vascular wall becoming rigid. 71
The pleiotropic cytokine transforming growth factor beta (TGF-β) is involved in many cellular activities, like migration, apoptosis, differentiation, and proliferation of cells. Because of an excessive TGF-β signaling, VSMCs from patients with Marfan Syndrome show an overactive RhoA/ROCK pathway. As was previously mentioned, these cells have higher levels of contractile protein markers expressed, which suggests that their cellular stiffness has risen. Furthermore, increased levels of transcription factor A linked to nuclear translocation of myosin were noted. These results suggest a mechanism wherein elevated TGF-β signaling eventually initiates RhoA/ROCK activation, which in turn causes greater stiffness. Through this mechanism, myosin-related transcription factor A may become more nuclearly localized, which may initiate a novel transcription program that changes the phenotype of VSMCs. 72 Nevertheless, this specific observation needs confirmation because of the contradictory results for RhoA/ROCK in VSMCs from Marfan syndrome patients.
By means of other processes involving polo-like kinase (Plk1), cullin-3, or neurolipin-2 (NLP-2), RhoA/ROCK signaling contributes in the stiffness of the VSMC. One piece of the gear that causes RhoA degradation is called cullin-3.
73
Increased arterial stiffness is the result of RhoA buildup caused by a ‘cullin-3 loss-of-function mutation (CUL3Δ9)’. In VSMCs, Ang II-mediated activation of RhoA/ROCK is regulated by Polo-like kinase 1 (Plk1) that is generally activated during cellular division. This results in the production of stress fibers, which effectively enhances cellular stiffness (Figure 6). Reduced vascular elasticity and an abnormal organization of cellular stress fibers are the results of specific deletion of Plk1 in VSMCs.
74
The diagram shows the molecular mechanisms contributing to vascular stiffness. It highlights several pathways that lead to the accumulation of RhoA, the activation of the RhoA/ROCK signaling pathway, and the transition of vascular smooth muscle cells (VSMC) to a stiffer phenotype. Key factors include mutations in Cullin-3, Ang II-mediated activation of RhoA/ROCK, and TGF-β signaling, which collectively increase vascular stiffness. This process is crucial in various cardiovascular conditions. Created with BioRender.com.
Furthermore, Plk1 controls RhoA’s Ang II-dependent activation, which puts Plk1 ahead of RhoA in the control of VSMC rigidity. RhoA/ROCK is downregulated in bladder SMC upon activation of the cellular receptor protein NRP2. As a result, there is a decrease in cellular stiffness because MLC is not as phosphorylated. Although this pathway is active in bladder SM, further research is required to determine if NRP2 signaling has any possible impact on cellular stiffness and VSMC phenotypic switching in particular. 75
7. Rho-kinase and vascular remodeling
RhoA/Rho-kinase signaling may be connected to vascular remodeling, which is a crucial aspect of vascular disorders. Normal development involves vascular remodeling, which takes part in a number of physiological functions. However, anatomical alterations to the vasculature can be both adaptive and pathogenic, resulting in the development of arterial disease and the aggravation of cardiovascular dysfunctions including hypertension and atherosclerosis. A powerful growth factor that contributes to the homeostasis of the artery wall is angiotensin II (Ang II). 76 Cardiac remodeling may be influenced by cardiac inflammation through activation of the cardiac angiotensin system. Data showing that Ang II regulates the cytoskeleton and stimulates the Rho-kinase pathway have directly linked Rho-kinase to pathologic vascular remodeling. 77 This is because Ang II is responsible for producing some of the cytoskeletal changes that arise during vascular remodeling, particularly in VSMCs. 78
7.1. Atherosclerosis
Additionally, vascular inflammation and atherosclerosis are facilitated by phosphorylated kinase. The interplay between severe inflammation and cholesterol buildup is what defines atherosclerosis. Selective Rho-kinase inhibitors cause a reduction in atherosclerotic plaque formation, a decrease in vascular inflammation, and an increase in eNOS. 79 Furthermore, studies employing ROCK1 (−/−) mice shown that ROCK1 in macrophages generated from bone marrow is essential for developing atherosclerosis, partly by mediating macrophage chemotaxis and foam cell production. 80 According to recent suggestions, statins, also known as HMG-CoA reductase inhibitors, approved as first-line treatments for hyperlipidemia to lower the risk of adverse cardiovascular events, have vascular benefits that are comparable to those of selective RhoA/Rho-kinase inhibitors in vitro 81 acting as an anti-atherogenic agent without lowering cholesterol. Both animal models’ cultured VSMCs and human samples showed this impact. 82 Additionally, Y-27632 decreased lesion growth in the apolipoprotein E knockout mice. 83
7.2. Hypertension
One of the most prevalent cardiovascular conditions, arterial hypertension is characterized by increased vascular contractility and changed vascular tone, which raise blood pressure. 84 It is accompanied by VSMC proliferation, migration, and variable degrees of arterial wall inflammation, all of which contribute to vascular remodeling. An essential function of the Rho-kinase pathway is to control arterial blood pressure. In 1997, it was discovered that Rho-kinase signaling plays a part in arterial hypertension. 85 In three experimental models of hypertension, a Rho-kinase inhibitor was found to lower arterial blood pressure in that research. Furthermore, Rho-kinase may potentially control blood flow directly by acting on the central nervous system or indirectly by interfering with the production and function of eNOS. 86 Both spontaneously hypertensive rats and hypertension patients have enhanced Rho-kinase pathways. Since several studies have shown that Rho-kinase inhibitors, including fasudil and Y-27632, may be useful treatments for hypertension, atherosclerosis, and other cardiovascular illnesses, this route has been extensively studied. 87 It has recently been shown that SAR407899, a new and powerful selective Rho-kinase inhibitor, has a better impact than Y-27632 and fasudil and shows promise antihypertensive efficacy. Still, a long way remains until it is therapeutically employed to treat hypertension. 88
7.3. Heart failure
Rho-kinase has a role in irreversible myocardial injury and regulates myofibrillar Ca+2 sensitivity in heart muscle. Rho-kinase is also implicated in the pathophysiology of cardiovascular remodeling 89 and by reducing the size of infarcts, which is the primary factor in the development of heart failure, its suppression is important in treating failing hearts. NOS and PI3 K/AKT activation are involved in Rho-kinase inhibition’s cardioprotective impact. 90 Nevertheless, future studies are required to determine the effectiveness of Rho-kinase inhibitor drugs across a broad dosage range, in vivo animal models that replicate the clinical context, and during different index ischemia times. 91 It has been proposed recently that pitavastatin, in particular, may enhance heart remodeling and function by producing eNOS linked to the Rho-kinase pathway. 92 Data demonstrating that statins reduce the intracellular amounts of other proteins, including RhoA, might help to explain this. Furthermore, statins inhibit RhoA activity and stop ROCK from activating, which can control the stability of eNOS mRNA.93,94
7.4. Vascular inflammation
Beyond its role in structural remodeling, accumulating evidence indicates that Rho-kinase also orchestrates vascular remodeling through inflammation-driven mechanisms. RhoA/ROCK signaling plays a central role in vascular inflammation by directly regulating pro-inflammatory cytokine production and transcriptional activation. Activation of the RhoA/ROCK axis stimulates NF-κB signaling, leading to increased expression of inflammatory cytokines such as TNF-α and IL-6 in both endothelial cells and vascular smooth muscle cells (VSMCs). 95 ROCK-dependent NF-κB activation further promotes the upregulation of endothelial adhesion molecules, including ICAM-1, VCAM-1, and E-selectin, thereby enhancing leukocyte adhesion, transmigration, and vascular inflammation. In VSMCs, RhoA/ROCK-mediated inflammatory signaling contributes to phenotypic switching toward a pro-inflammatory state, amplifying cytokine release and extracellular matrix remodeling. 96 Collectively, these inflammatory mechanisms establish RhoA/ROCK signaling as a critical molecular bridge linking endothelial dysfunction, immune activation, and chronic vascular inflammation.
8. Current and potential therapeutic uses of RhoA/ROCK inhibition
8.1. Established clinical uses
Since 1994, the RhoA/ROCK inhibitor fasudil hydrochloride has been clinically approved for the treatment of cerebral vasospasm. 97 Beyond this indication, fasudil has also demonstrated therapeutic efficacy in coronary vasospasm, pulmonary hypertension, and glaucoma, highlighting the broad vascular relevance of RhoA/ROCK signaling. Although the pleiotropic actions of Rho/ROCK across multiple cell types raise theoretical safety concerns, this widespread activity may also partially explain the acceptable clinical safety profile observed with fasudil. In addition, ROCK inhibition has been proposed as a potential therapeutic strategy for cardiovascular complications associated with sepsis, diabetic macular edema, Raynaud’s phenomenon, and multiple sclerosis. 98
8.2. Limitations and safety concerns
Despite promising clinical and experimental outcomes, several limitations hinder the widespread use of RhoA/ROCK inhibitors. Given the involvement of Rho/ROCK signaling in multiple organ systems, systemic adverse effects remain a concern. Importantly, teratogenicity represents a major safety issue, as Rho-kinase has been shown to play a critical role in fetal cardiac development, limiting use during pregnancy.23,99
Another major challenge is the lack of isoform selectivity of many existing inhibitors, which may suppress other kinases in addition to ROCK, thereby increasing the risk of off-target effects during long-term or high-dose therapy. Furthermore, differential inhibition of ROCK1 versus ROCK2 may result in distinct pharmacological consequences, emphasizing the need for isoform-specific therapeutic strategies. 100
8.3. Novel inhibitors and advanced delivery systems
In recent years, advances in 3D molecular design and structure-based virtual screening have enabled the identification of novel Rho/ROCK inhibitors with improved potency and selectivity. These in silico approaches allow millions of compounds to be screened from chemical databases, followed by experimental validation of promising candidates. 101 Compared with first-generation inhibitors such as fasudil, several newly developed molecules—including benzodioxane derivatives, 2,3-diaminopyrazine derivatives, and benzyl pyridinethiazole-based amide compounds—exhibit enhanced selectivity for ROCK kinases and reduced activity toward unrelated kinases such as protein kinase A and C. 102 Notably, some of these compounds display preferential inhibition of ROCK2, supporting isoform-targeted therapeutic development.
To further reduce systemic toxicity, a long-acting hypoxia-activated prodrug of fasudil has recently been developed. This prodrug demonstrates selective activation within hypoxic pulmonary tissue in pulmonary hypertension and within cancer cells, thereby minimizing off-target effects while preserving therapeutic efficacy. 103
In parallel, vasculature-targeted delivery strategies have emerged as a promising approach to enhance tissue specificity. In a leporine carotid artery model, stents eluting rhosin (a RhoA inhibitor) significantly reduced neointimal hyperplasia compared with bare metal stents. 104 Similarly, anagliptin treatment attenuated intimal hyperplasia following balloon injury in mice through inhibition of the SOD-1/RhoA/JNK pathway, further supporting the role of RhoA/ROCK inhibition in limiting VSMC-driven vascular injury. 105
More recently, a multi-drug electrospun external vascular sheath has been developed to prevent venous graft failure. This system enables time-dependent release of fasudil dihydrochloride, everolimus, and simvastatin directly at the graft site. 106 Fasudil promotes early endothelial protection and repair, everolimus suppresses mid-phase VSMC proliferation, and simvastatin reduces late-stage atherosclerotic changes. 107 Collectively, these tailored delivery platforms highlight the growing potential of localized and isoform-selective ROCK inhibition as a future strategy for managing complex cardiovascular diseases.
9. Limitations of current ROCK inhibitors and emerging strategies
Despite the widespread use of fasudil and Y-27632 as prototypical ROCK inhibitors, both compounds have significant pharmacokinetic and translational limitations. Fasudil exhibits a short plasma half-life (∼30 minutes) and requires intravenous administration due to poor oral bioavailability, restricting its long-term therapeutic applicability. 108 Similarly, Y-27632 demonstrates limited metabolic stability and lacks suitability for clinical use because of its poor bioavailability and rapid systemic clearance. A major limitation shared by both agents is their lack of tissue specificity, resulting in widespread inhibition of ROCK isoforms across multiple organ systems, which increases the risk of off-target effects.
To overcome these barriers, ongoing research is exploring novel strategies such as nanoparticle-mediated delivery, which enhances vascular targeting and prolongs drug circulation time, as well as vasculature-targeted prodrug formulations designed to improve site-specific activation and reduce systemic exposure. 109 These advancements aim to improve pharmacokinetic profiles, tissue selectivity, and overall therapeutic potential of next-generation ROCK inhibitors.
10. Discussion
With a focus on its function in inflammation-mediated cardiovascular pathology, this study offers a thorough description of the RhoA/ROCK signaling pathway in vascular dysfunction. The current review emphasizes new data that connects RhoA/ROCK activation to inflammatory signaling, oxidative stress, and endothelial dysfunction, whereas earlier research has mostly concentrated on the function of ROCK in vascular contraction and remodeling. This review highlights the interaction between vascular inflammation and RhoA/ROCK signaling and explores possible treatment approaches that target this pathway by incorporating new experimental and clinical data. This viewpoint clarifies the wider pathogenic significance of RhoA/ROCK signaling and suggests possible directions for further study in metabolic and cardiovascular vascular diseases.
This narrative review has some limitations even though it offers a comprehensive summary of current knowledge. The selection of studies may be impacted by the availability of published literature and possible publication bias because the review is not predicated on a systematic search method. Furthermore, there is still a dearth of extensive clinical data about the long-term therapeutic efficacy and safety of ROCK inhibitors, and the review mainly concentrates on experimental and preclinical findings. Therefore, to better identify the translational potential of targeting the RhoA/ROCK pathway in vascular disorders, more thorough research and carefully planned clinical studies are needed.
11. Conclusion
The RhoA/ROCK signaling pathway plays a central role in regulating vascular tone, endothelial function, and vascular remodeling in cardiovascular diseases. Its overactivation promotes oxidative stress, nitric oxide depletion, vascular smooth muscle cell phenotypic switching, and inflammation, contributing to conditions such as atherosclerosis, hypertension, and heart failure. Although pharmacological ROCK inhibition has shown promising vascular protective effects, challenges including limited isoform selectivity and potential off-target effects remain. Emerging evidence suggests that combining ROCK inhibition with complementary approaches such as AMPK activation or antioxidant therapy may provide enhanced vascular protection by targeting multiple interconnected mechanisms. Such dual-pathway strategies may be particularly beneficial in diabetic vascular complications and warrant further investigation for improved therapeutic outcomes.
Footnotes
Acknowledgements
The authors acknowledge the use of Artificial Intelligence (AI) tools (e.g., ChatGPT, OpenAI) solely for the purpose of language refinement, grammar correction, and improvement of the overall readability of the manuscript. No AI tools were used for the generation, analysis, or interpretation of scientific data, nor for the creation or modification of results, figures, or conclusions. All authors confirm that they have carefully reviewed and verified the content of the manuscript and take full responsibility for its accuracy, originality, and scientific integrity.
Author contributions
Kuldeep Singh contributed to conceptualization, literature review, manuscript drafting, and final editing. Divya Jain contributed to literature curation, data interpretation, and manuscript writing. Arpan Kumar Tripathi was involved in data collection, figure conceptualization, and manuscript revision. Touseef Begum contributed to critical review of scientific content and editing. Mohammad Muztaba assisted in literature analysis and drafting support. Shamim contributed to data interpretation and manuscript refinement. Jeetendra Kumar Gupta provided supervision, validation, and critical revision of the manuscript. Abdullah Al Noman, as the corresponding author, provided conceptual oversight, manuscript review, and final approval. Mukesh Chandra Sharma contributed to technical review and content validation. Pranab Dev Sharma contributed to scientific editing, proofreading, and final manuscript approval. All authors have read and approved the final version of the manuscript.
Funding
The authors further declare that no funds, grants, or other support from any Govt. or other private funding agency has been received during the preparation of this manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article, as no datasets were generated or analysed during the current study.