
Abstract
Sphingolipids form a broad class of lipids with diverse functions ranging from membrane constituents to intracellular second messengers and extracellular mediators. They can be rapidly generated or converted into each other and they play pivotal roles in various cellular processes, many of which are broadly associated with inflammation and apoptosis. Among the numerous sphingolipids, ceramide and sphingosine-1-phosphate (S1P) have received the greatest attention. Ceramide is a hydrophobic molecule that is increased in the lungs of patients with cystic fibrosis and chronic obstructive pulmonary disease (COPD). Ceramide is the eponym for ceramide-rich membrane platforms. that need to form as a prerequisite to the uptake of several microorganisms including Pseudomonas aeruginosa, and as a prerequisite to many signaling processes including apoptosis and increased vascular permeability. Accordingly, abnormal amounts of enzymes involved in the synthesis of ceramide, such as neutral or acid sphingomyelinase, are found in emphysematic smokers and in patients with severe sepsis, and are considered as novel pharmacological targets. S1P acts as an extracellular mediator that opposes several actions of ceramide and acts by binding to G-protein coupled S1P receptors (S1P1–S1P5). Of particular interest are S1P1 receptors that enhance vascular barrier functions and are antiapoptotic. Therefore, S1P1-receptor ligands are suggested as novel drugs for COPD and acute lung injury. S1P is a potent chemotaxin for many leukocytes, it organizes lymphocyte trafficking and is involved in several key symptoms of asthma such as airway hyperresponsiveness and pulmonary eosinophil sequestration. S1P is formed by sphingosine kinases that have been identified as possible drug targets for the treatment of asthma. Based on these findings, several new drugs have recently been developed to specifically target sphingomyelinases, sphingosine kinases and S1P receptors for the treatment of COPD, cystic fibrosis, asthma and acute lung injury.
Keywords
Introduction
Sphingolipids represent a large group of lipid molecules. The prefix ‘sphingo-’ refers to the sphinx-like, enigmatic, behavior of this group of lipids. By some accounts, the ancient riddle of the sphinx was: ‘There are two sisters: one gives birth to the other and she, in turn, gives birth to the first’ [Grimal, 1996, p. 324]. To some extent, this old riddle outlines the modern view on sphingolipids, namely that of membrane sphingolipids giving rise to a large class of mediators, and that of sphingolipid mediators giving birth to numerous cell functions including alterations of the cell membrane. Today, the functions of sphingolipids in the regulation of fundamental biological processes (e.g. apoptosis, immunity, vascular homeostasis) have become so vast and diverse that no single review can hope to cover it all. Our purpose here is to review the emerging significance of sphingolipids, in particular, ceramide and sphingosine-1-phosphate (S1P) for the pathophysiology of several respiratory disorders, that is, Niemann–Pick disease, acute lung injury (ALI), asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF). The respective chapters contain two parts: the first part discusses the relevant biological processes regulated by sphingolipids and the second part therapeutic interventions and the first clinical based on these insights.
A brief account of sphingolipid biology
Sphingolipids are amphipathic molecules with variable hydrophobic and hydrophilic properties (Figure 1). The hydrophobic part comprises a so-called sphingoid base (usually 18 carbons in length, e.g. sphingosine or sphinganine) that is linked to a fatty acid through an amide bond [Dickson et al. 2002]. The hydrophilic part consists of just hydroxyl groups in the simplest sphingolipids, whereas the more complex sphingolipids have phosphates and sugar residues attached. There are at least five different sphingoid bases present in mammalian cells with more than 20 arrangements of fatty acids with different length of the alkyl chain and different degrees of saturation and hydroxylation [Fuller, 2010; Futerman and Hannun, 2004]. Sphingolipids are synthesized in a pathway that begins in the endoplasmic reticulum and ends in the Golgi apparatus, but most of these lipids are enriched in the plasma membrane and in endosomes where they perform many of their biological functions [Uhlig and Gulbins, 2008; Van Meer et al. 2002].
Overview over the metabolism of sphingolipids and their major functions. For further details see text. PKC, protein kinase C; PP!, protein phosphatases 1; PP2A, protein phosphatases 2A; S1P, sphingosine-1-phosphate; SMase, sphingomyelinase.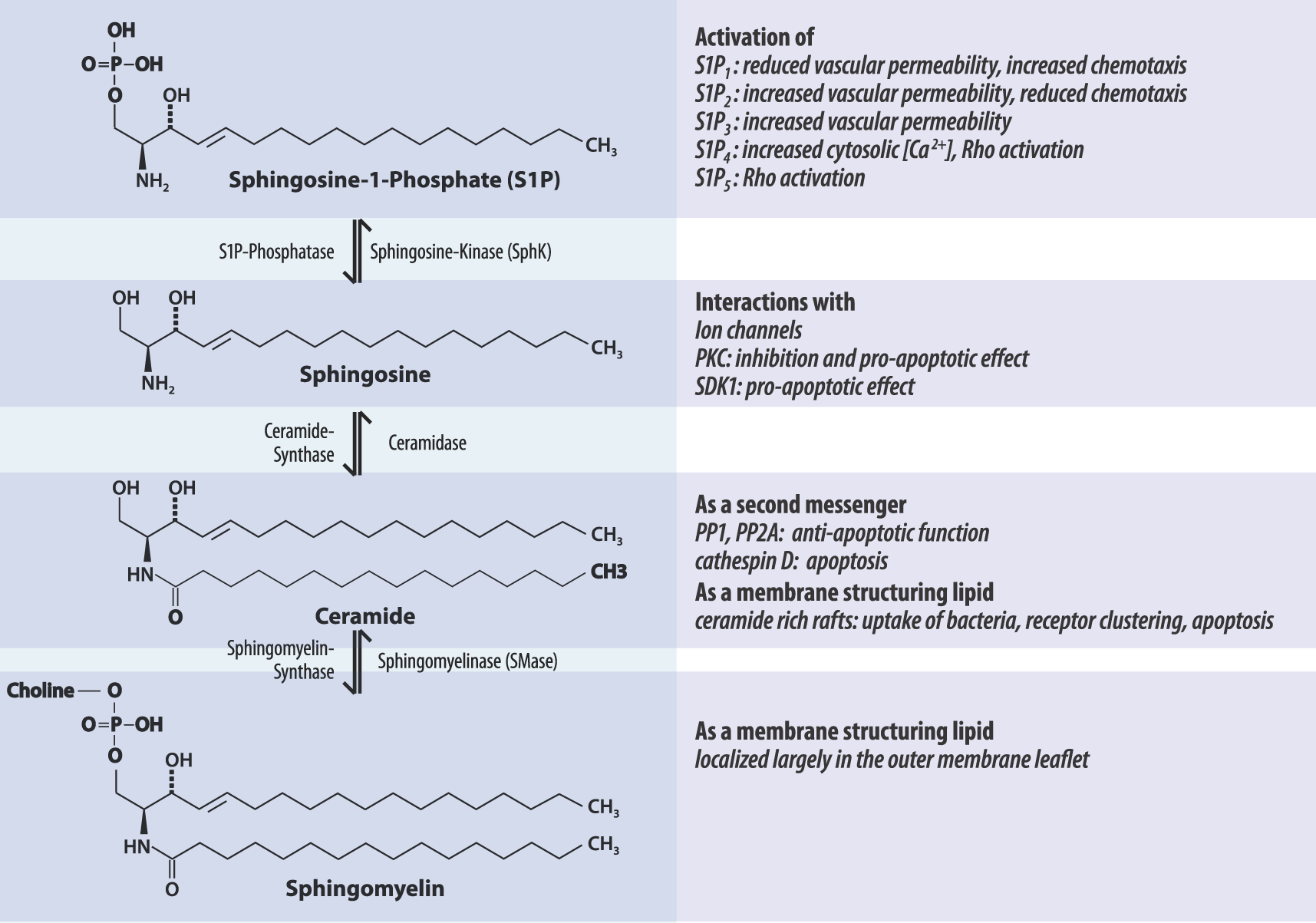
Sphingosine is the backbone of sphingolipids, whose phosphorylation generates S1P, and whose acylation generates ceramide; most studies to date have focused on these two molecules. Interestingly, these two lipids or their receptors often assume antagonistic roles, as in the regulation of vascular permeability, apoptosis or chemotaxis (Figure 1). Furthermore, it seems likely (just as with the eicosanoids) that new sphingolipids with unexpected functions will continue to be discovered. Recent examples are dihydrosphingosine-1-phosphate, sphingosylphosphorylcholine that reduced lung injury caused by endotoxemia [Murch et al. 2008], or ceramide-1-phosphate that may regulate the survival and apoptosis of the lung cancer cells [Tada et al. 2010; Mitra et al. 2007].
Sphingomyelinase isoenzymes catalyze the breakdown of sphingomyelin by cleavage of the phosphorylcholine linkage, thereby producing ceramide. Sphingomyelinases are characterized by their pH optimum as acid, neutral and alkaline isoforms [Marchesini and Hannun, 2004; Duan et al. 2003; Chatterjee et al. 1999; Quintern et al. 1989]. The acid sphingomyelinases (ASMases) are classified into a lysosomal L-SMase and a secretory S-SMase, which are both derived from the same gene (SMPD1) [Uhlig and Gulbins, 2008; Schissel et al. 1998]. The L-SMase requires mannose-6-phosphate receptors for its trafficking into lysosomes (Figure 2), interacts with lipid membranes via its sphingolipid activator domain, and catalyzes sphingolipid turnover and degradation [Ni and Morales, 2006]. ASMase activation is localized on the outer membrane leaflet [Koval and Pagano, 1991], and is often involved in signaling processes [Blachnio-Zabielska et al. 2010; Ussher et al. 2010; Gulbins et al. 1997; Zhang et al. 1997; Dobrowsky and Hannun, 1993]. ASM can translocate to the plasma membrane either rapidly [Yang et al. 2010; Jin et al. 2008] or slowly [Jenkins et al. 2010]. The slow (probably also constitutive) pathway depends on serine residue 508 and brings the S-SMase to the cell surface [Jenkins et al. 2010]. The rapid mechanism has been explained by fusion of lysosomes containing pre-formed L-SMase with the plasma membrane [Xia et al. 2010; Jin et al. 2008]; the exact molecular identity of this form of ASMase has not yet been established. In tumor necrosis factor (TNF)-receptor endosomes, the 72 kDa ASM is processed by the sequential action of caspase 7 and caspase 8 into an enzymatically active 57 kDa form [Tchikov et al. 2010]. Notably, the rapid translocation of ASMase to the plasma membrane can be blocked by the induction of lysosomal proteases through cyclic antidepressants such as imipramine [Jenkins et al. 2010; Yang et al. 2010].
Proposed pathway for the maturation and translocation of acid sphingomyelinase (ASMase). A slow pathway for translocation is induced by tumor necrosis factor (TNF) (upper left), a rapid pathway by platelet-activating factor (PAF) and the Fas ligand (Fas-L). Inhibitors are shown in blue, stimulating agents in red. Part of this figure has been adapted from Jenkins et al. [2010].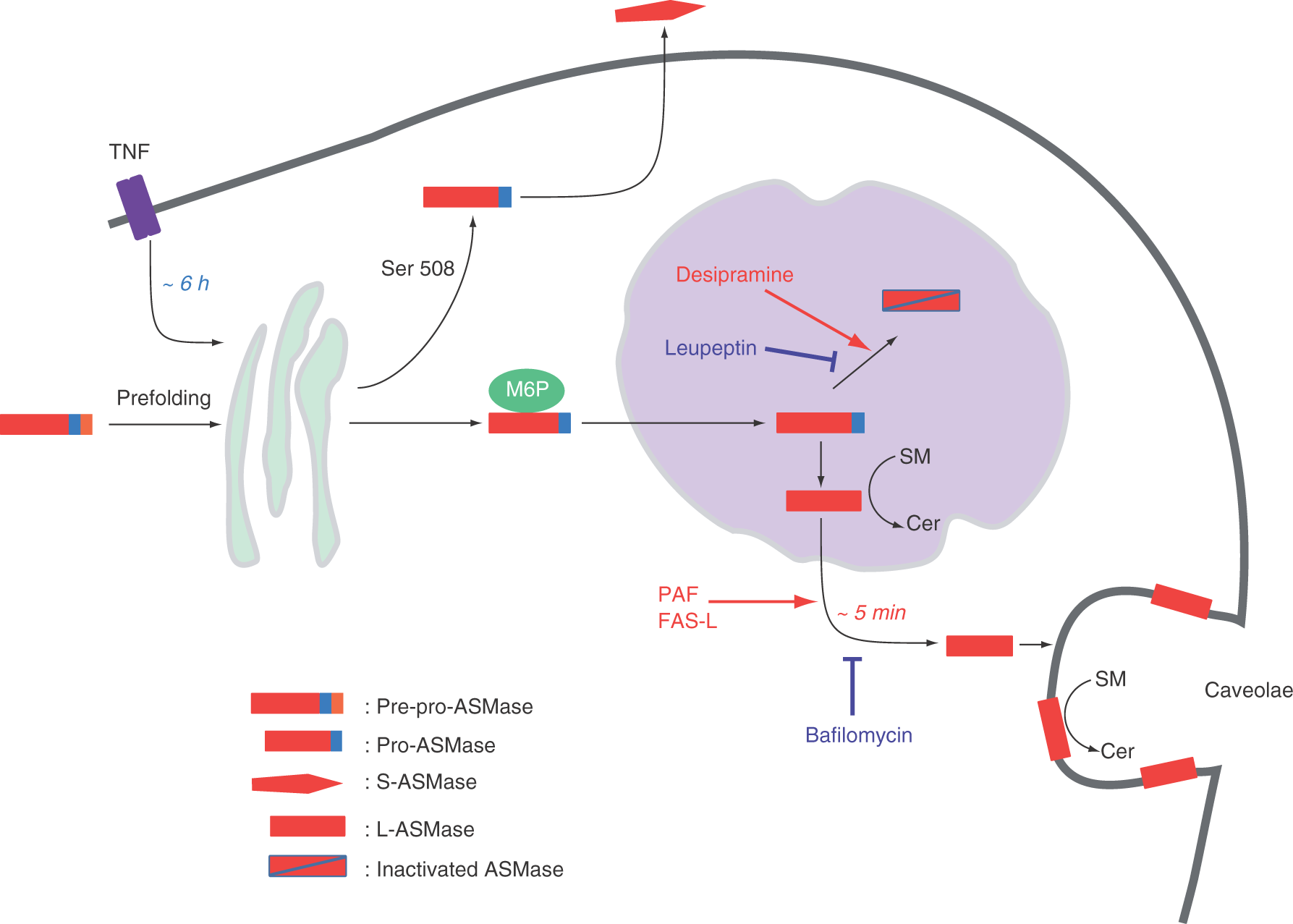
From the cell surface the extracellular ASMase isoforms can further translocate to the extracellular space by yet poorly defined mechanisms. Extracellular ASMase activity is present in most body fluids [Takahashi et al. 2000] and increased levels are found in several respiratory disorders [Yang et al. 2010; Von Bismarck et al. 2008; Petrache et al. 2005; Goggel et al. 2004; Grassme et al. 2003, 1997], although the function of this circulating ASM is unknown. Endothelial cells are considered as the main source of circulating ASMase [Marathe et al. 1998].
Neutral sphingomyelinase (NSMase) is activated by several stress inductors such as TNF, shear stress [Czarny et al. 2004], oxidative stress [Castillo et al. 2007] and cigarette smoke [Filosto et al. 2010; Levy et al. 2009] that frequently lead to apoptosis. NSMase can translocate to the membrane dependent on p38 [Clarke et al. 2007] and protein kinase C-delta (PKCδ) [Clarke et al. 2008]. Activation of p38 is also responsible for the NSMase-dependent activation of cyclooxygenase-2 by peptidoglycans in macrophages [Chen et al. 2009]. Finally, in neutrophils NSMase appears to be required for the proper organization of the leading edge when orientating towards a chemotaxin [Sitrin et al. 2010]. Recent studies have identified NSMase as a target in COPD caused by cigarette smoking [Filosto et al. 2010].
Niemann-Pick disease (NPD, type A, B and C) is a rare genetic disorder that is caused by A-SMase deficiency. Both type A and B forms are inherited as recessive traits, and both are due to mutations in the SMPD1 gene [Mcgovern et al. 2008; Simonaro et al. 2002]. Type A NPD is the infantile form, characterized by neurological deficits, enlarged livers and spleens, pulmonary infections and death by age 2–3. In contrast, type B NPD is the later onset form, in which patients exhibit little or no neurological symptoms but may have severe and progressive visceral organ abnormalities, including hepatosplenomegaly, pulmonary insufficiency, and cardiovascular disease [Schuchman, 2009]. Type C NPD is a complex lipid storage disorder cause by defects in cholesterol trafficking [Guillemot et al. 2007]. Impaired pulmonary function is a major clinical finding in patients with type B NPD [Guillemot et al. 2007; Mendelson et al. 2006] and is also observed in ASMase knockout mice [Dhami et al. 2001]. Patients with NPD suffer from symptoms of COPD and other respiratory problems [Guillemot et al. 2007]. A bronchoalveolar lavage study on young patients with NPD showed an abnormal percentage of polymorphonuclear cells and the presence of the Niemann-pick cells: ‘foamy’ macrophages that are shaped by the intracellular accumulation of sphingomyelin or unesterified cholesterol [Guillemot et al. 2007].
Ceramide exists in tissue in many different forms with variable fatty acid chain length. Unlike the sphingoid precursors, ceramide is not water-soluble and is located in membranes where it participates in raft formation [Liu et al., 1995, Gulbins et al., 2003]. The physical properties of ceramide ensure that ceramides are concentrated preferentially into lateral liquid-ordered microdomains. ceramides could segregate into distinct, high-temperature melting ceramide-enriched domains: ceramide-rich platforms [Jin et al., 2008, Stancevic et al., 2010]. Ceramide is generated within these rafts by the action of ASMase and merges into larger formations modifying the membrane structure and its biophysical properties [Jin et al. 2008; Gulbins et al. 2004; Gulbins and Kolesnick, 2003; Liu and Anderson, 1995]. Specific receptor molecules and signaling proteins cluster within such platforms, which initiate and greatly amplify the primary signals [Uhlig and Gulbins, 2008].
Ceramide can be produced de novo or during the metabolism of other sphingolipids (Figure 1), usually by the activation of the sphingomyelinases (see above) or ceramide synthase [Marchesini and Hannun, 2004; Okino et al. 2003]. Many agonists including chemotherapeutic agents, TNF, platelet-activating factor (PAF), 1,25-dihydroxy-vitamin D3, endotoxin, interleukins, nerve growth factor, irradiation and heat shock stimulate hydrolysis of sphingomyelin to produce ceramide [Lang et al. 2005; Goggel et al. 2004; Gulbins and Grassme, 2002; Huwiler et al. 2000; Kolesnick et al. 1994]. Apart from its role in cellular membranes, ceramide may act as a second messenger: it interacts with cathepsin D in TNF-receptor endosomes [Tchikov et al. 2010], as well as with ceramide-activated protein phosphatases and ceramide-activated protein kinases to regulate glycogen synthesis and insulin resistance in response to apoptotic stimuli [Blachnio-Zabielska et al. 2010; Ussher et al. 2010; Zhang et al. 1997; Dobrowsky and Hannun, 1993]. In addition, ceramide can interact with ion channels and trigger pore formation [Siskind et al. 2002; Gulbins et al. 1997].
S1P, a zwitterionic lysophospholipid, is an important cellular metabolite, derived from sphingosine. S1P is synthesized by two sphingosine kinases (SphK1, SphK2) and degraded by S1P phosphatases, lipid phosphate phosphatases or S1P lyase [Spiegel and Milstien, 2003]. Sphingosine kinases phosphorylate sphingosine to form S1P. Two mammalian isoforms of sphingosine kinases have been identified, named SphK1 and SphK2, which display different catalytic properties, subcellular locations and substrate specificity [Spiegel and Milstien, 2003]. SphK is expressed in bronchial epithelium and airway smooth muscle cells [Nishiuma et al. 2008]. Apparently, SphK knockout mice develop an adaptive phenotype, which makes studies with these mice inconclusive [Puneet et al. 2010]. SphK activity can be stimulated by a variety of stimuli such as muscarinic M2 receptor agonists, activation of immunoglobulin E (IgE) receptors, histamine, growth factors and various cytokines [Oskeritzian et al. 2008; Alemany et al. 2007; Taha et al. 2006; Pfaff et al. 2005; Choi et al. 1996].
S1P is a potent messenger molecule which plays an ubiquitous regulatory role in cell proliferation and survival, angiogenesis and endothelial barrier functions, cellular Ca2+ homeostasis, cell contacts and adhesion, chemotaxis and cytoskeletal organization [Alemany et al. 2007; Spiegel et al. 2003; Watterson et al. 2003]. S1P performs its extracellular effects through specific G protein-coupled receptors on cell surfaces, designated S1P1 to S1P5 [Watterson et al. 2003]. Different S1P receptor subtypes may act antagonistically: endothelial barrier is enhanced by S1P1 but impaired by S1P2 and S1P3; chemotaxis is stimulated by S1P1 but inhibited by S1P2 [Uhlig et al. 2008; Spiegel et al. 2002]. S1P is also an intracellular second messenger, where it may have effects even opposite to those of its extracellular counterpart [Itagaki et al. 2007].
Vascular permeability and acute lung injury
The altered permeability of the pulmonary endothelial barrier for water and proteins leads pulmonary edema formation. This is a characteristic hallmark of inflammatory responses and contributes to the morbidity and mortality in anaphylaxis, sepsis and acute lung injury (ALI) or in its most severe form to the acute respiratory distress syndrome (ARDS) [Ware et al., 2000, Dhillon et al., 2005].
Mechanisms of vascular permeability
Sphingolipids play critical and varied roles in the regulation of vascular permeability – both in homeostasis and disease. Key areas of regulation by sphingolipids are the intracellular levels of endothelial calcium and NO, and the endothelial cytoskeleton. One fundamental finding in recent years relates to the barrier-stabilizing properties of S1P1 receptors [Singleton et al. 2006, 2005]. These receptors experience tonic activation by the relatively high serum S1P levels of 0.5 µM, which are probably derived from erythrocytes and possibly endothelial cells [de et al. 2010; Kim et al. 2009; Marsolais et al. 2009; Berdyshev et al. 2005]. If these receptors are blocked in healthy animals, the consequence is pulmonary edema [Sanna et al. 2006]. Another homeostatic control mechanism of the endothelium may be the upregulation of nitric oxide (NO) production in response to increased shear stress that is mediated by the NSMase [Czarny et al. 2004]. We would like to emphasize that the pulmonary vascular bed differs in many important aspects from systemic vessels and thus findings in other extrapulmonary tissues may not always apply to the lung [Kübler et al. 2010]. Hence in the following we will consider mainly studies in the pulmonary endothelium.
Endothelial calcium plays a critical role in the regulation of pulmonary vascular permeability by activation of endothelial myosin light chain kinase, the regulation of cadherins, PKCα and RhoA GTPase [Komarova and Malik, 2010]. In human pulmonary artery endothelial cells, S1P causes only a very brief and transient (2 min) increase in intracellular calcium that is unrelated to the barrier-stabilizing properties of S1P [Camp et al. 2009]. ASMase and ceramide, however, appear to be involved in the calcium-dependent pulmonary edema that is caused by PAF [Goggel and Uhlig, 2005]. Interestingly, the PAF-induced increase in intracellular calcium levels is blocked by imipramine and is replicated by perfusion with ASMase or ceramide [Samapati et al., 2009]. We currently examine the hypothesis that ceramide may activate TRP-C channels (canonical transient receptor potential channels).
Endothelial NO synthase (eNOS) production regulates pulmonary vascular permeability which is increased both in eNOS-deficient mice [Predescu et al. 2005] and by the NOS inhibitor L-NAME [Yang et al. 2010]. Remarkably, however, despite increased permeability both conditions do not lead to edema formation, indicating that reduced endothelial NO levels do facilitate edema formation triggered by other mechanisms, but do not cause it [Kübler et al. 2010]. This is borne out by recent studies showing that part of the PAF-induced edema is caused by impairing endothelial NO production [Yang et al. 2010]. Importantly, the rapid (within minutes) cessation of endothelial NO production in response to PAF is mediated by ASMase and ceramide [Yang et al. 2010]. The authors suggest that this is explained by the formation of ceramide-rich caveolae that recruit caveolin-1 into these newly formed membrane domains, which interacts with eNOS and traps it inactive in the membrane (Figure 3) [Yang et al. 2010; Parton and Simons, 2007; Li et al. 2006]. In contrast to ASMase, NSMase and S1P activate eNOS and increase endothelial NO levels in pulmonary endothelial cells in culture [Thomas et al. 2005; Czarny et al. 2004]. S1P3 can activate eNOS and provoke NO production in vascular endothelial cells through Gi- and Akt-mediated phosphorylation of eNOS in concert with a Gq-mediated, Ca2+/calmodulin-dependent mechanism [Nofer et al. 2004]. It is unknown how this affects pulmonary vascular permeability.
Proposed pathway for the formation of ceramide-rich caveolae in response to platelet-activating factor (PAF). As described in Figure 2, PAF leads to the rapid translocation of acid sphingomyelinase (ASMase) to the plasma membrane. In endothelial cells, this translocation appears to occur preferentially into caveolae, where the formation of ceramide leads to further recruitment of caveolin-1 (Cav-1), which is well known to interact and thus inhibit with endothelial nitric oxide synthase (eNOS). Therefore, cav-1 and eNOS accumulate in caveolae, with the consequence that endothelial nitric oxide production stops. This hypothesis is explained in greater detail by Yang and colleagues and Kübler and colleagues [Kübler et al. 2010; Yang et al. 2010].
The endothelial cytoskeleton and stress fiber formation appear closely related to edema formation as numerous, mostly in vitro, studies have demonstrated [Vandenbroucke et al. 2008]. The enhancement of endothelial barrier by S1P is mediated by S1P1, a Gi-coupled receptor, which leads to activation of the Rac GTPase and subsequent cytoskeletal reorganization and focal adhesion assembly [Mcverry et al. 2005]. This process involves many other proteins such as Tiam1, cortactin, α-actinin, paxillin, focal adhesion kinase, and the GPCR kinase-interacting proteins GIT1 and GIT2 [Dudek et al. 2004; Shikata et al. 2003] and caveolin-rich microdomains [Singleton et al. 2005]. Activation of S1P2 and S1P3 receptors, however, inhibits Rac through activation of Rho-kinase, stimulation of the 3′-specific phosphoinositide phosphatase PTEN [Inoki et al. 2006; Sanchez et al. 2005], and weakening of junctional integrity [Sanchez et al. 2007]. Thus, depending on the S1P receptor subtypes and their localization, and probably also depending on its concentration, S1P can tip the balance of pulmonary endothelial permeability in both ways.
S1P1 and S1P3 can also be transactivated. In human pulmonary or umbilical vein endothelial cells, S1P1 receptors are transactivated by protein C receptors, protease activated receptor-1 receptors and CD44s, leading to endothelial barrier enhancement [Singleton et al. 2006; Feistritzer et al. 2005; Finigan et al. 2005], whereas S1P3 receptors are transactivated by CD44v10 and μ-opioid receptors leading to increased endothelial permeability [Singleton et al. 2007, 2006].
Treatment of acute lung injury
ALI is a severe disorder with a mortality of greater than 25% and a proven causative pharmacological treatment is not available. A hallmark of this disease is endothelial and epithelial barrier dysfunction [Ware and Matthay, 2000]. From the mechanisms discussed above it is obvious that sphingolipids could affect this disorder in several ways: ALI could be caused by the activation of ASM or by abnormal S1P levels. In addition, pharmacological activation of S1P1 receptors seems attractive as a treatment for ALI.
The importance of ASMase for the development of ALI has been shown in many models such as pulmonary edema caused by PAF [Kerlin and East, 1992], lipopolysaccharide (LPS) [Claus et al. 2005; Goggel et al. 2004; Haimovitz-Friedman et al. 1997], acid instillation [Goggel et al. 2004] or repeated lung lavage [von Bismarck et al. 2008]. Importantly, impairment of ASMase improved not only vascular barrier functions (as shown in all studies above), but also oxygenation [von Bismarck et al. 2008; Goggel et al. 2004]. The studies above used ASM knockout mice or indirect rather unspecific inhibitors of the ASM pathway such as the xanthogenate D609 or tricyclic antidepressants such as imipramine (for mode of action, see Figure 2). For therapeutic purposes new specific ASMase inhibitors will be needed, such as a class of bisphosphonates that was recently discovered [Roth et al. 2009]. Given the extracellular location of ASMase, such inhibitors may not need to be cell permeable.
Pulmonary disorders, sphingolipids and selected interventions.

ASMase, acid sphingomyelinase; NSMase2, neutral sphingomyelinase-2; S1P, sphingosine-1-phosphate; SphK, sphingosine kinase.
Abnormal S1P levels could cause pulmonary edema when S1P levels are either too low so that S1P1 receptors are not sufficiently activated or too high so that S1P2 or S1P3 receptors become activated. Unfortunately, S1P levels have not yet been reported in models of ALI or in patients with ALI. It has been shown that instillation of high doses (0.5 mg/kg) of S1P or SEW-2871 (S1P1-receptor agonist) causes mild pulmonary edema when given alone, at a dose that was also able to reduce LPS-induced edema [Sammani et al. 2010]. Interestingly, when lung injury was elicited by intratracheal LPS administration inactivation of S1P2 or S1P3 receptors partially protected against protein hyperpermeability [Sammani et al. 2010], suggesting that these two receptors may contribute to pulmonary barrier dysfunction in sepsis.
S1P1-receptor agonists may be valuable pharmacological drugs to treat ALI. Both S1P and selective S1P1-receptor agonists (SEW-2871) have been effective in reducing pulmonary edema and/or neutrophil infiltration in lung injury caused by LPS, high tidal volume ventilation, ischemia reperfusion or necrotizing pancreatitis [Sammani et al. 2010; Mcverry et al. 2004; Peng et al. 2004]. FTY720, a synthetic derivative of the fungal product myrocin, bears a strong resemblance to sphingosine and S1P. It was originally considered a specific S1P1-receptor agonist with barrier-enhancing properties lacking untoward effects on S1P3 receptors [Sammani et al. 2010], airway smooth muscle cells [Rosenfeldt et al. 2003] and cardiac toxicity [Forrest et al. 2004; Hale et al. 2004]. In line with this, FTY720 exhibits barrier-enhancing functions both in vitro and in vivo [Dudek et al. 2007; Peng et al. 2004; Sanchez et al. 2003]. The exact mechanism of FTY720 barrier-enhancing function remains unclear because FTY appears to be internalized and to downregulate S1P1-receptor signaling instead of activating this pathway. It was reported that FTY720 potently enhances endothelial barrier function partially via a S1P1-independent mechanism that involves an alternative Gi-coupled receptor [Dudek et al. 2007]. These findings may offer additional insight into barrier-regulatory pathways of FTY720 that may serve as useful clinical targets for modulation of vascular leak.
Sphingosine-kinase 1 is the predominant isoform responsible for the presence of S1P in the circulation [Venkataraman et al. 2006]. SphK1 is believed to act through S1P to modulate severe inflammatory signaling cascade like phosphoinositide 3-kinase, nuclear factor κB in macrophages and inflammatory signaling in neutrophils. Increased permeability of endothelial cells challenged by thrombin and activated polymorphonuclear neutrophils (PMNs) can be attenuated by treatment with SphK inhibitors [Itagaki et al. 2010]. Sphk1 inhibition also allows modulation of inflammation and inhibition of PMN activation via control of [Ca2+]i [Lee et al. 2004]. SphK1 is upregulated in stimulated human phagocytes and intraperitoneal phagocytes of sepsis patients [Puneet et al. 2010]. Pharmacological blockade of SphK1 with siRNA or with a novel pharmacological agent impaired phagocyte production of endotoxin-induced proinflammatory cytokines and protected against lung injury mortality in animal models of sepsis LPS [Puneet et al. 2010]. Importantly, the SphK inhibitor was effective even when given after induction of sepsis and acted synergistically with an antibiotic [Puneet et al. 2010].
Allergic responses and asthma
Asthma is characterized by airway remodeling, dysfunctions of the bronchial epithelium and airway hyperresponsiveness caused by chronic T helper 2 (Th2)-type inflammation [Uhlig and Martin, 2008; Magnussen, 2003]. Sphingolipids play critical roles in several of the fundamental processes involved in this disease. For instance, administration of S1P causes production of Th2 cytokines such as IL-4 and IL-13 [Roviezzo et al. 2010]. Notably, S1P may also direct the immune response toward Th17 responses [Roviezzo et al. 2010; Liao et al. 2007].
Mechanisms of allergic disease
This section looks at airway smooth muscle and airway remodeling. S1P receptors are present on smooth muscle cells and regulate many of their functions [Watterson et al. 2005]. S1P triggers S1P2 receptors on airway smooth muscles in a Rho-dependent manner causing airway hyperresponsiveness and under certain conditions also airway smooth muscle contraction [Chiba et al. 2010; Roviezzo et al. 2010; Szczepaniak et al. 2010; Kume et al. 2007; Rosenfeldt et al. 2003]. Remarkably, exogenously administered S1P itself can produce airway hyperresponsiveness in naive mice [Roviezzo et al. 2010, 2007; Haberberger et al. 2009; Nishiuma et al. 2008; Kume et al. 2007] and conversely, ovalbumin-sensitized mice show airway hyperresponsiveness to S1P [Roviezzo et al. 2007]. In addition, S1P is involved in cholinergic airway contraction because M2 receptor-induced bronchoconstriction is mediated by sphingosine kinase and S1P [Pfaff et al. 2005]. With respect to airway smooth muscle, it has been suggested that the S1P/SPK pathway plays a major role in pathological but not in physiological conditions [Roviezzo et al. 2007].
S1P can also contribute to airway remodeling: S1P promotes the proliferation and potentiates the growth of human airway smooth muscle cells [Ammit et al. 2001; Ediger et al. 2000] and fibroblasts [Desai et al. 1992; Zhang et al. 1991]. Furthermore, S1P produced by SphK1 has recently been shown to be involved in fibroblast differentiation into myofibroblasts, which was believed to be relevant to lamina reticularis thickening [Batra et al. 2004]. In addition, it was shown that transforming growth factor-β (TGF-β) stimulates SphK1 that in turn released S1P that activates S1P2 and S1P3, leading to α-smooth muscle actin expression in lung fibroblasts, which is hallmark of their differentiation into myofibroblasts [Batra et al. 2004]. Another characteristic of allergic airways is altered mucus production that may also be partly regulated by S1P [Kono et al. 2010].
Mast cells play a central role in the development of asthma. Cross-linking of their high-affinity IgE receptor FcεRI induces activation of sphingosine kinases, which represents an early critical signal for the allergen-induced rise in mast cell calcium [Jolly et al. 2004; Melendez et al. 2002; Choi et al. 1996]. In fact, the initial calcium wave is thought to depend entirely on S1P, whereas the second wave depends on IP3 (reviewed by Melendez [2008]). S1P can be exported from mast cells by ABC transporters to amplify mast cell responses by binding to S1P receptors. Activation of S1P2 is required for mast cell degranulation while S1P1 activation is important for their migration to sites of inflammation. S1P binding to S1P1 induces mast cell chemotaxis, cytoskeletal rearrangements and triggers mast cell migration towards an antigen gradient [Olivera and Rivera, 2005; Jolly et al. 2004], a mechanism by which low antigen concentrations could attract mast cells to the action site via S1P1. S1P2, however, mediates mast cell activation and promotes degranulation [Jolly et al. 2004]. S1P secretion from mast cells also provokes inflammation by activating and recruiting other immune cells like eosinophils [Roviezzo et al. 2004] and Th2 lymphocytes [Sawicka et al. 2003].
Chemotaxis of most inflammatory cells including mast cells, eosinophils and lymphocytes is regulated by S1P [Rivera et al. 2008]. For instance, lungs isolated from S1P-treated mice displayed an increase in mast cell numbers [Roviezzo et al. 2010]. Of note, the effect of S1P may depend on the concentration of S1P, such that low concentrations promote and high concentrations block it (reviewed by Rivera and colleagues [Rivera et al. 2008]); a bell shaped concentration–response curve is typical for many chemokines [Ludwig et al. 1997]. Of great importance is the finding that S1P is a critical signal for the homing of immune cells to lymphoid organs and for regulating their egress into blood and lymph [Matloubian et al. 2004].
Treatment of asthma
Taken together, there is ample evidence that sphingolipids, above all S1P, regulate many parts of the allergic cascade. Therefore, several studies have examined the abundance of S1P in asthma, the expression and genetic variation of S1P receptors and the therapeutic effects SphK inhibitors or S1P-receptor ligands.
S1P levels are elevated in the airways of people with asthma after segmental allergen challenge [Ammit et al. 2001] and also in allergic mice [Nishiuma et al. 2008]. S1P1 receptors are downregulated in human asthma and people with gene variants in this receptor are more prone to asthma [Sun et al. 2010]. In contrast, SphK1 and SphK2 activity was upregulated in the ovalbumin-sensitized mouse asthma model [Nishiuma et al. 2008; Roviezzo et al. 2007]. Other changes that have been described in mouse allergy models are downregulation of S1P2 receptors [Chiba et al. 2010a] and upregulation of S1P phosphatase 2 [Haberberger et al. 2009], and S1P lyase [Haberberger et al. 2009]. Thus, the alterations in sphingolipid-related genes and proteins in asthma appear highly complex.
Novel pharmacologic interventions in asthma are of great mechanistic and therapeutic interest. Recently, it was demonstrated in ovalbumin-sensitized mice that intratracheal administration of a novel S1P1-receptor agonist (CYM-5442) prevented release of chemokines (CCL5, CCL17), the accumulation of dendritic cells and CD4+ lymphocytes in draining lymph nodes, and finally the eosinophil and lymphocyte accumulation in the airways [Marsolais et al. 2011].
Several studies have examined inhibitors of sphingosine kinase. Among these treatments nebulization of N,N-dimethylsphingosine (DMS) and intranasal SphK1 siRNA proved particularly effective in that they attenuated all aspects of allergic responses in ovalbumin-sensitized mice: IgE production, airway hyperreactivity, production of Th2 cytokines, eosinophil and lymphocyte infiltration and mucus production [Lai et al. 2008; Nishiuma et al. 2008]. Other SphK inhibitors such as 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SK-I) [Nishiuma et al. 2008] or 4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol (SKI-II) [Chiba et al. 2010b] were less effective: they prevented airway hyperresponsiveness and pulmonary eosinophil sequestration, but had no or only small effects on Th2 cytokines and IgE production. Similar effects as with SK-I and SKI-II were seen in sensitized SphK1-deficient mice that lack the airway hyperresponsiveness and eosinophilic infiltration, but show unaltered Th2 cytokines [Haberberger et al. 2009]. This latter work, however, also issued a note of caution with respect to the use of sphingosine kinase inhibitors for the treatment of asthma, because the SphK-deficient mice developed more severe vascular remodeling than the wild type mice [Haberberger et al. 2009]. All these data demonstrate a pivotal role of SphK in airway hyperresponsiveness and eosinophil infiltration in allergic airway disease. The regulation of IgE production and Th2 responses, however, was not blocked in all studies, and the role of SphK in this area needs further study.
The role of ceramide in cystic fibrosis and pulmonary infections
CF is a chronic condition caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene [Kerem et al. 1989; Rommens et al. 1989]. These genetic defects of CFTR cause pulmonary symptoms like chronic lung inflammation and frequent and chronic infections of the lung with P. aeruginosa, but also with Staphylococcus aureus, Burkholderia cepacia and Haemophilus influenzae [Becker et al. 2010a]. The mechanisms that mediate chronic lung inflammation in patients with CF are unknown. However, an increasing number of studies suggest that sphingolipids play an important regulatory role in CF with respect to pulmonary infections and inflammation.
Mechanisms of ceramide-rich platforms and pH-dependent ceramide formation
Ceramide-enriched membrane platforms are believed to play an important regulatory role in CF and pulmonary infections. Ceramide is generated within lipid rafts by the action of ASMase, and causes small rafts to merge into larger platforms [Jin et al. 2008; Gulbins et al. 2004; Gulbins and Kolesnick, 2003; Liu and Anderson, 1995]. These ceramide-enriched membrane domains are thought to permit the spatial and temporal organization of receptors and signaling molecules. It was reported that CD95 [Cremesti et al. 2001; Grassme et al. 2001a, 2001b], CD40 [Grassme et al. 2002], CD14 [Pfeiffer et al. 2001], CFTR [Kowalski et al. 2004] and caspase 8 [Eramo et al. 2004] may trigger the formation of such ceramide-enriched membrane platforms, cluster in or interact within it. NF-κB was also shown to be upregulated by the ASM–ceramide axis [Gill and Windebank, 2000], and destruction of ceramide-enriched platform prevented nuclear translocation of NF-κB in respiratory epithelial cells and cellular apoptosis [Kowalski et al. 2004; Grassme et al. 2003]. Hence, it was suggested that CFTR expression controls the accumulation of membrane ceramide [Bodas et al. 2011]. Finally, CFTR has also been described as a transporter of S1P [Boujaoude et al. 2001].
Particularly relevant to CF are the findings that both CFTR and CD95 receptors cluster within ceramide-rich platforms upon infection [Grassme et al. 2003, 2000], because CD95 mediates apoptosis of cells infected with P. aeruginosa and because CFTR can act as a receptor for P. aeruginosa that mediates the internalization of the bacteria [Pier et al. 1997, 1996]. Of note, airway epithelial cells in CD95-deficient animals failed to undergo apoptosis and showed a high susceptibility to P. aeruginosa infections [Grassme et al. 2000], suggesting that apoptosis is central in the coordinated defense against pulmonary infections with P. aeruginosa.
An imbalance between ASMase and acid ceramidase caused by elevated pH was observed in the airways of Cftr-deficient mice [Teichgraber et al. 2008] and in their alveolar macrophages [Zhang et al. 2010]. It was suggested that lack of CFTR leads to alkalinization of intracellular vesicles, most likely in secretory lysosomes, with immediate consequences for ceramide synthesis: up to a pH of 6.0 acid ceramidase activity is inhibited, whereas ASMase activity still remains above 60% [Teichgraber et al. 2008; He et al. 2003]. This imbalance favors accumulation of ceramide that can trigger chronic pulmonary inflammation, death of respiratory epithelial cells and DNA deposition in bronchi.
Treatment of cystic fibrosis and pulmonary infection
The fundamental mechanisms discussed above suggest that the pathophysiology associated with CF may be at least partly corrected by interfering with ceramide formation in lysosomes and ceramide-rich platforms, and with its role in the immune defense against P. aeruginoasa and other microorganisms.
Lungs from patients with CF show increased expression levels of pulmonary and tracheal ceramide [Becker et al. 2010b; Yu et al. 2009; Teichgraber et al. 2008], in particular of the species C16:0, C18:0 but not C22:0 [Brodlie et al., 2010]. In addition, CFTR-deficient mice accumulated ceramide in the pulmonary epithelium [Teichgraber et al., 2008, Brodlie et al., 2010, Bodas et al., 2011]. Also inhibition of membrane-ceramide release by amitriptyline showed protective effects in controlling P. aeruginosa-LPS-induced lung injury in Cftr(-/-) mice compared with that in Cftr(+/+) mice, indicating that membrane-CFTR is required for controlling lipid-raft ceramide levels [Bodas et al., 2011]. ASMase inhibition in lungs of Cftr-deficient mice normalized the concentrations of the proinflammatory mediators IL-1 and IL-8, as well as the number of macrophages and neutrophils [Teichgraber et al. 2008]. In line with this, patients with CF showed a correlation between ceramide-containing cells and neutrophils [Brodlie et al. 2010]. Bronchial epithelial cell death and DNA depositions containing plugs were also prevented by pharmacological or genetic inhibition of ASMase in Cftr-deficient mice [Teichgraber et al. 2008]. ASMase inhibition as a therapeutic strategy for treating patients with CF has already been examined in small clinical studies: lung functions were improved and upper respiratory tract infections were reduced in patients with CF treated with amitriptyline [Becker et al. 2010b; Riethmuller et al. 2009].
Pulmonary infections trigger rapid ASMase activation and formation of ceramide-enriched membrane platforms [Grassme et al. 2003], which are crucial for the internalization of pathogens. This has been demonstrated for infections with Neisseria gonorrhoeae [Grassme et al. 1997], Escherichia coli, S. aureus, Listeria monocytogenes, Salmonella typhimurium and P. aeruginosa [Mccollister et al. 2007; Falcone et al. 2004; Utermohlen et al. 2003; Esen et al. 2001; Crouch Brewer et al. 1996]. Interestingly, administration of galactosyl ceramide led to improved clearance of P. aeruginosa from infected lungs [Nieuwenhuis et al. 2002]. It was also found that infection of ASMase-deficient mice with P. aeruginosa caused a cytokine storm and finally death of the mice [Grassme et al. 2003]. Another study showed down-regulation of A-SMase and reduced ceramide levels in Ps. aeruginosa-infected CFTR knockout mice together with augmented inflammation [Yu et al., 2009]. All these findings demonstrate that ceramide-enriched membrane platforms are crucially involved in the proper response to pulmonary infections, including adequate cytokine responses.
Ceramide-rich platforms thus seem to play a critical role in many of the pathophysiological manifestations of CF. Other proteins that interact with ceramide platforms and play a critical role in apoptosis on P. aeruginosa infection are the major vault protein (MVP) [Dudez et al. 2008; Kannan et al. 2008; Kowalski et al. 2007; Kannan et al. 2006] and the Src-like kinase Lyn [Kannan et al. 2008, 2006]. With the help of Lyn-deficient mast cells, Lyn was shown to be necessary for the internalization of P. aeruginosa and regulating inflammatory cytokines [Kannan et al. 2006]. MVP was essential for optimal epithelial cell internalization and clearance of P. aeruginosa. Infection of MVP-deficient mice led to internalization failure of the pathogens into lung epithelial cells and an increase in mortality [Kowalski et al. 2007].
Taken together, the role of ceramide in CF is janus headed. On the one hand it is required to successfully fight infection with P. aeruginosa; on the other hand its overexpression in CFTR-deficient mice and in patients with CF apparently contributes to the pathopyhsiological alterations seen in that disease. The complex task therefore may be to therapeutically adjust the pulmonary ceramide levels to their physiological range.
Apoptosis and emphysema
Emphysema is defined by destruction of alveolar walls causing abnormal permanent enlargement of the distal airspaces ultimately leading to impaired oxygenation. Emphysema is part of COPD primarily caused by cigarette smoking, and currently there is no effective treatment to halt or to repair the alveolar wall enlargement in emphysema [Uhlig and Martin, 2008; National Heart, Lung, and Blood Institute, 1985]. The pathogenesis of emphysema is thought to be related to chronic lung inflammation and it has been suggested that alveolar cell apoptosis and oxidative stress, among other things, are critical in the pathogenesis of emphysema [Tuder et al. 2003].
Mechanisms of apoptosis and oxidative stress
Ceramide appears to be one of the crucial mediators of alveolar destruction in emphysema by mediating oxidative stress and apoptosis of alveolar endothelial and epithelial cells [Petrache et al. 2008, 2005]. Ceramide may mediate apoptosis by a variety of mechanisms, such as activation of kinase suppressors [Zhang et al. 1997], protein phosphatases 1 and 2A [Chalfant et al. 2002], cathepsin D [Heinrich et al. 2000], or alteration of plasma membrane properties and clustering signaling molecules by forming ceramide-rich platforms [Gulbins and Grassme, 2002]. S1P as a downstream product of ceramide metabolism and contrary to ceramide mediates cell survival and proliferation [Cuvillier et al. 1996]. It is believed that a balance between ceramide and S1P levels regulates cellular survival and homeostasis [Payne et al. 2002; Morita et al. 2000]; sometimes this is called the sphingolipid rheostat. Increased ceramide levels leading to apoptosis may result from de novo synthesis or activation of SMase isoenzymes [Medler et al. 2008].
Reactive oxygen species regulate various cellular functions, including apoptosis and cell proliferation [Dumitru et al. 2007; Goldkorn et al. 2005]. Ceramide is causing oxidative stress, in part by altering the mitochondrial potential and in part by downregulation of antioxidative defense such as superoxide dismutase [Petrache et al. 2008; Andrieu-Abadie et al. 2001]. Conversely, oxidative stress can activate ASMase via a redox mechanism resulting in release of ceramide and formation of ceramide-enriched platforms that serve to cluster DR5 and CD95, which further regulate apoptosis [Dumitru et al. 2007; Dumitru and Gulbins, 2006]. Oxidative stress-induced ceramide accumulation finally leads to activation of caspase-3 and apoptotic cell death [Goldkorn et al. 2005]. In addition, NSMase can become activated by redox mechanisms, and the following ceramide generation was inhibited by pretreatment antioxidants [Dumitru et al. 2007; Goldkorn et al. 2005].
Treatment of emphysema
Patients with emphysema show increased ceramide levels that were localized to alveolar septal cells and macrophages [Brodlie et al. 2010; Petrache et al. 2005]. Ceramide is formed in response to cigarette smoke, for example after 4 weeks of intermittent exposure of 5 h a day and 5 days a week [Petrache et al. 2008]. While ASMase activity may be increased [Petrache et al. 2008], in the case of cigarette smoke ceramide and subsequent apoptosis appear to be generated by neutral sphingomyelinase 2 [Filosto et al. 2010; Castillo et al. 2007]. Mice that were heterozygous for NSMase2 demonstrated a significant decrease in ceramide generation after cigarette smoke exposure, while ASMase knockout mice maintained wild type ceramide levels, indicating that NSMase2 rather than ASMase is activated by cigarette smoke exposure [Filosto et al. 2010]. These effects were abrogated by treatment with the antioxidant N-acetyl cysteine or with anti-NSMase2 siRNA. Importantly, lung tissue from humans with emphysema (smokers) expressed higher levels of NSMase2 [Filosto et al. 2010].
Pulmonary emphysema may result from rarification of pulmonary vessels [Giordano et al. 2008], a process in which endothelial ceramide formation appears to play a pivotal role. This was convincingly demonstrated in emphysema induced by vascular endothelial growth factor blockade, which leads to ceramide formation in alveolar cells by the de novo pathway and feedforward activation of the ASMase [Petrache et al. 2005]. In this model, the increased ceramide generation was critical to trigger alveolar cell death, oxidative stress and emphysema. Neutralization of ceramide or inhibition of its synthesis prevented emphysema [Petrache et al. 2005]. In line with the concept of a cellular ceramide-S1P rheostat, ceramide formation, apoptosis and emphysema were all attenuated by treatment with exogenous S1P [Diab et al. 2010; Petrache et al. 2005]. In addition, the S1P1 agonist SEW2871 showed significant protective effects on airspace enlargement concomitant with attenuation of vascular endothelial growth factor receptor inhibitor-induced lung apoptosis in pulmonary endothelial cells [Diab et al. 2010].
Recently, it was shown that the decreased clearance of apoptotic cells (efferocytosis) by alveolar macrophages may also contribute to lung inflammation in pulmonary emphysema [Petrusca et al. 2010]. It was demonstrated that sphingosine, derived from ceramide via ceramidase, impaired the alveolar macrophages clearance of apoptotic cells via a mechanism involved sphingosine generation and Rac1 modulation [Petrusca et al. 2010].
Taken together, there is ample evidence to show that high ceramide levels are characteristic for COPD and that this is probably related to endothelial and epithelial cell apoptosis. Studies in animal models suggest that interference with ceramide synthesis may offer novel therapeutic approaches for this debilitating disease.
Summary
Sphingolipids are increasingly recognized as critical mediators in major pulmonary diseases (Table 1). The properties of ceramide which are most relevant here are the organization of inflammatory responses and apoptosis. Many of the diverse actions of ceramide can be explained in terms of ceramide-rich rafts or caveolae that provide critical signaling platforms in the regulation of vascular permeability, the recognition and uptake of bacteria and in apoptosis. Given these diverse functions, inhibition of ceramide synthesis is expected to have multiple effects. However, probably because ceramide can be generated by several different enzymes and pathways, it may be possible to block ceramide synthesis without intolerable side effects. This is illustrated by the well established use of tricyclic antidepressants that also reduce ASMase activity. Inhibitors of ASMase have been suggested as treatments for CF and ALI. In addition, altered ceramide metabolism may provide a link between CF and COPD, two diseases that not only share a critical role of P. aeruginosa infections but possibly also alterations in CFTR expression [Bodas et al. 2011]. The other major sphingolipid that has received much attention is S1P that is critically involved in leukocyte chemotaxis, lymphocyte trafficking, vascular permeability and cell survival. As such, it plays a pivotal role in the development of asthma and in this disease antagonists directed against S1P synthesis or receptors have been effective in preclinical models. On the other hand, activation of the S1P1 receptor increases survival and vascular permeability and therefore S1P1 agonists may be useful new drugs in the treatment of ALI and COPD.
The emerging importance of sphingolipids in respiratory disorders is reflected in the recent development of novel and increasingly specific pharmacological agents in this area, such as ASMase inhibitors [Arenz, 2010; Roth et al. 2009], S1P1 agonists [Marsolais et al. 2011, 2009], or SphK1 inhibitors [Puneet et al. 2010]. The evolving knowledge in this field gives rise to the hope that such drugs may provide novel therapeutic options in the treatment of several respiratory disorders.
Footnotes
Funding
Supported by the Deutsche Forschungsgemeinschaft as the project Uh 88/8-2 within the SPP1267 ‘Sphingolipids’.
Conflicts of interest statement
None declared.