
Abstract
The rapid expansion of novel technologies in cancer research over the past several years has led to a dramatically improved understanding of the molecular biology of lung cancer. As a consequence, novel targeted therapies are rapidly being developed. In this review, we summarize the most important molecular pathways in lung cancer and describe the clinical evidence for the development of therapies against these targets.
Introduction
With an estimated 222,000 new cases and an estimated 157,000 deaths in the United States for the year 2010, lung cancer remains the most deadly malignancy and accounts for as many deaths as colon, breast, and prostate cancers combined [Jemal et al. 2010]. Lung-cancer-specific survival rates, however, have improved slightly since the 1990s [Owonikoko, 2010], which gives us reason to hope that better understanding of the underlying molecular etiology of lung cancer and the development of better treatment approaches including targeted therapies will have a growing clinical impact. In this review, we describe the most relevant molecular and biochemical pathway alterations in lung cancer and review the clinical evidence of novel therapies aimed at those molecular targets.
Introduction to signaling pathways
Normal mammalian cells require growth factor stimulation to maintain cellular homeostasis. Growth factors are essential for the cell to take up nutrients from the environment, migrate, and to proliferate or undergo apoptosis. Normal epithelial cells also exhibit contact-dependent inhibition, limiting cellular proliferation when in contact with other cells. Cellular homeostasis and the balance between proliferation, inhibition, and apoptosis are usually tightly regulated. In contrast, cancer cells overcome this growth factor dependency, and contact inhibition through the acquisition of genetic and epigenetic changes that ultimately lead to oncogene activation and tumor-suppressor-gene inactivation. As the technology for genome-wide sequencing and epigenetic analysis has evolved rapidly over recent years, so has our understanding of the molecular and biochemical pathways underlying lung cancer.
The major goal of targeted therapies is the identification and therapeutic exploitation of an ‘Achilles’ heel’ which renders the tumor particularly vulnerable to a particular treatment. Several concepts have been developed to highlight the underlying molecular mechanisms. In ‘oncogene addiction’ [Weinstein et al. 1997], in which the tumor depends on the activation of a single oncogene, targeted therapy specifically against this oncogene will result in cancer regression. In human studies, this concept has first been proven for chronic myelogenous leukemia (CML) and the targeting of the bcr-abl fusion gene by imatinib, but has also been proven for epithelial growth factor receptor (EGFR)-mutated lung cancers. ‘Synthetic lethality’ describes a concept in which inactivation of one particular gene may confer a survival advantage, but inactivation of a second gene in a synthetically lethal relationship would result in cell death [Tucker and Fields, 2003]. This principle has been proven for BRCA-mutated breast cancer, where targeting PARP1 with small molecule inhibitors results in dramatic clinical responses [Fong et al. 2009]. In the majority of lung cancers it is currently unknown whether there is such a molecular Achilles’ heel. It is possible that the majority of molecular targets are merely components of pathways that are co-activated; therefore, therapies against these targets could result in synergistic effects with other targeted or conventional cancer therapies such as chemotherapy and radiotherapy rather than as a single agent alone.
Molecular pathways of lung cancer
EGFR–Ras–Raf–MAPK pathway
Activating mutations in the EGFR or its downstream targets k-Ras and b-Raf highlight the importance of this pathway for the development of non-small-cell lung carcinoma (NSCLC). Mouse models with inducible EGFR mutations [Ji et al. 2006] or k-Ras mutations [Fisher et al. 2001] show that these are sufficient to induce lung cancer and to lead to rapid regression when the oncogenic stimulus is withdrawn. In line with these observations, the initial treatment experience with the EGFR tyrosine kinase inhibitor (TKI) gefitinib in patients with EGFR-mutated lung cancer showed unprecedented responses [Lynch et al. 2004; Paez et al. 2004]. Activating EGFR mutations occur in about 8–10% of all patients with NSCLC, but are more frequent in never or light smokers, women and people of Asian descent. In this subgroup, the incidence of EGFR mutations approaches 40–50% [Bell et al. 2005]. African Americans, on the other hand, seem to have a significantly lower incidence of EGFR mutations [Leidner et al. 2009]. Normally, EGFR signals downstream through both the Ras–Raf–Mek and Erk (Figure 1A) pathways as well as through activation of PIK3, AKT and MTOR (Figure 1B). K-ras mutations, which occur in about 20–30% of all lung adenocarcinomas in both smokers and nonsmokers at even frequencies [Riely et al. 2008] are associated with a low likelihood of response to EGFR TKIs [Pao et al. 2005b] but not, counterintuitively, to the monoclonal antibody against EGFR, cetuximab [Khambata-Ford et al. 2010]. Mutations in b-raf are rare and occur in only about 2–3% of adenocarcinomas [Ueda et al. 2008; Brose et al. 2002]. Even in patients with EGFR mutations, resistance to EGFR TKI usually emerges after a period of time. Common resistance mechanisms include the emergence of the EGFR-T790M mutation [Pao et al. 2005a], which occurs in up to 50% of acquired TKI resistance and amplification of the c-met proto-oncogene [Engelman et al. 2007]. The T790M mutation does not interfere with the binding of the TKI to EGFR, but restores EGFR’s ability to also bind adenosine triphosphate (ATP) in the catalytic domain despite the presence of a TKI [Yun et al. 2008].
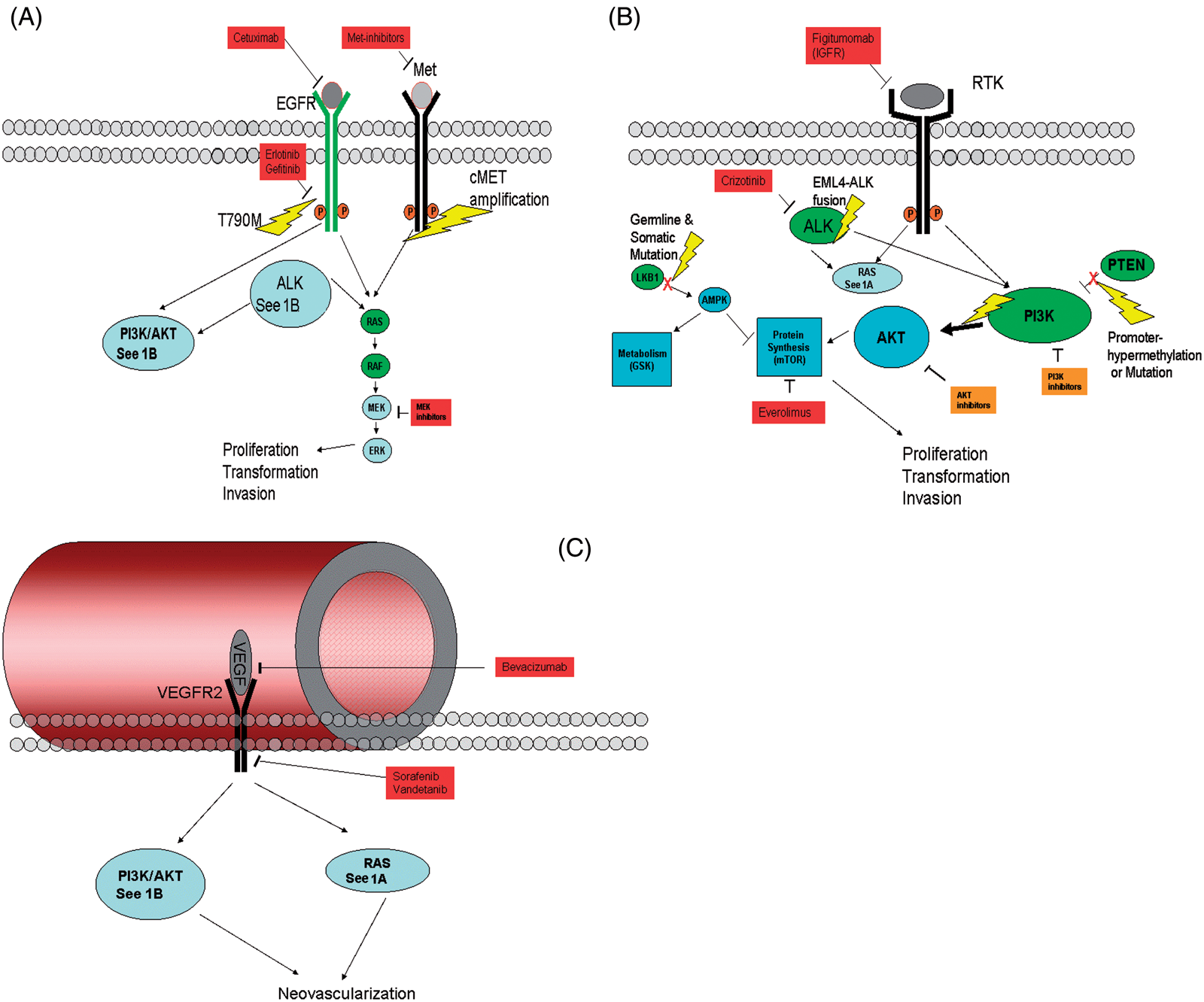
(A) Epithelial growth factor receptor (EGFR) signaling through the Ras–Raf–Mek–Erk pathway. EGFR is targeted by various tyrosine kinase inhibitors. Point mutation of EGFR to EGFR-T790M confers resistance to inhibitors. Amplification of the cMET gene also provides resistance to targeted therapies. (B) Receptor tyrosine kinases (including EGFR and insulin-like growth factor receptor [IGFR]) signal through Ras and PI3K–Akt–mTOR. Anaplastic lymphoma kinase (Alk) also signals through these pathways. The EML4–Alk fusion present in about 5% of adenocarcinomas constitutively activates these pathways. The protein PTEN suppresses activation of PI3K but is frequently inactivated in non-small-cell lung carcinoma (NSCLC) by mutation or promoter hypermethylation. Mutations in PI3KCa cause constitutive activation of the pathway. Mutation and epigenetic silencing effectors of the mTOR pathway (including the wnt axis component LKB1) induces hyperproliferation, transformation and invasive oncogenic phenotypes. Downstream, inhibition of the mTOR pathway by everolimus has been shown to be efficacious in renal cell carcinoma (RCC) and is currently being tested for activity in NSCLC. (C) Neovascularization promoted by VEGF signaling through Ras and Akt pathways is suppressed by small-molecule inhibitors sorefenib and vandetanib and monoclonal antibody drug bevacizumab.
PI3K–Akt–mTOR pathway
It has been estimated that Akt is one of the most frequently activated protein kinases in human cancer. The Phosphatidyl-inositol-3 Kinase (PI3K)–Akt–mammalian Target of Rapamycin (mTOR) pathway (Figure 1B) acts downstream from receptor tyrosine kinases such as EGFR, IGFR, cMET and ERBB3. Activation of the Akt pathway through PI3K activation has been recognized as a major mechanism of acquired resistance to EGFR TKIs in NSCLC. Two physiologic inhibitors provide additional checks and balances in this pathway. PTEN inhibits PI3K and acts upstream from Akt. Phosphate and tensin homolog PTEN is frequently inactivated in NSCLC either by mutations or promoter hypermethylation. Mutations of the catalytic subunit PI3KCa leads to constitutive activation. Mutations of PI3KCa in NSCLC are relatively rare (∼4%), but overexpression is a frequent event [Yamamoto et al. 2008]. mTOR signaling is also controlled by the tuberous sclerosis complex genes TSC1 and TSC2. The inhibitory effects of the TSC on mTOR are in part mediated by the LKB1 signaling cascade which activates TSC1 and TSC2 through AMP-activated kinase (AMPK) signaling. LKB1 germline mutations lead to the Peutz Jaegher’s syndrome, a condition characterized by an increased incidence of benign and malignant tumors, including lung cancer. With an incidence of about 20–30% in adenocarcinoma and about 50% in squamous cell carcinoma of the lung, LKB1 mutations are among the most frequent genetic aberrations in NSCLC. In mouse models, there is a strong synergistic interaction between LKB1 and k-ras mutations favoring carcinogenesis.
Vascular endothelial growth factor and angiogenesis
According to Judah Folkman’s famous ‘angiogenic switch hypothesis’ tumor growth and proliferation can only occur in the presence of tumor-induced neovascularization [Folkman et al. 1971]. Twenty years after his groundbreaking discovery that a tumor-derived angiogenesis factor (TAF) in fact induces neovascularization in a rat model, the vascular endothelial growth factor (VEGF) was cloned and characterized [Keck et al. 1989; Leung et al. 1989]. VEGF signals through several receptors, where VEGFR2 (Figure 1C) is mostly responsible for its pro-angiogenic properties. VEGFR2 homodimerizes upon engagement by VEGF transducing signals through the Erk, PI3K, and MAPK pathways, and thereby enhancing endothelial cell proliferation, vascular permeability and endothelial cell migration.
IGFR signaling
The insulin-like growth factor receptor IGF-1R is a member of the insulin receptor class of transmembrane receptor tyrosine kinases (Figure 1B). Its alpha subunit is the extracellular portion of the receptor, while the beta-subunit is the transmembrane and intracellular portion with the catalytically active site. IGF-1 is the most active ligand for IGF-1R, leading to homodimerization and phosphorylation. Akt is the major downstream target. Other ligands for the IGF-1R are insulin and IGF-2. The normal function of IGF-2 is as a growth promoting hormone during gestation. Its expression is tightly regulated in normal cells through genomic imprinting. Lung cancers show a frequent loss of imprinting with upregulation of IGF-2. By heterodimerizing with EGFR, IGF-1R signaling has also been associated with acquired resistance to EGFR inhibitors [Morgillo et al. 2007, 2006].
Epigenetic control of gene regulation
Under normal circumstances, epigenetic mechanisms of gene regulation play a major role in stem cell maintenance and the imprinting of the second female X chromosome. In lung cancer, it is well established that epigenetic events occur early on in carcinogenesis [McCabe et al. 2009; Herman and Baylin, 2003; Belinsky et al. 2002]. Silencing of tumor suppressor genes by methylation of CpG islands in the promoter regions of genes or alterations of the histone code have been well established in lung cancer for critical cell cycle genes such as p16 [Belinsky et al. 1998], DNA repair genes such as MGMT and hMLH-1, apoptosis inducers such as DAPK and genes involved in the Ras–(RASSF1) [Licchesi et al. 2008a] and the wnt signaling pathways (SFRP 1, 2, 4, 5, APC, LKB1) [Licchesi et al. 2008b]. In a cell culture model, siRNA knockdown of DNMT1 prevented smoke carcinogen-induced transformation of normal human bronchial epithelial cells and combined pharmacologic inhibition of DNA methyltransferases (DNMTs) and histone deacetylases (HDACs) prevented the formation of murine lung cancer [Damiani et al. 2008].
EML4–Alk translocation
Recent evidence showed that the anaplastic lymphoma kinase (Alk) can be aberrantly expressed under the control of the EML4-promoter in a small subset of NSCLC [Soda et al. 2007]. This occurs in about 5% of adenocarcinomas, but is more frequent in younger patients and those with minimal to no smoking history [Shaw et al. 2009]. Overamplified Alk signals through both the Ras–Raf–Erk (Figure 1A) pathways as well as through the PI3K–Akt–mTOR (Figure 1B) pathway and is being stabilized by HSP90 [Chen et al. 2010].
Targeted therapies in clinical practice
EGFR inhibitors: historical overview
EGFR inhibitors were developed in the early 2000s based on the observation that EGFR expression and overexpression occurs in the majority of NSCLC. Initial nonrandomized phase II clinical trials with the EGFR inhibitor gefitinib, dosed at both 250 mg and 500 mg per day in the second- and third-line setting, were encouraging and led to US Food and Drug Administration (FDA) accelerated approval in 2003 [Fukuoka et al. 2003; Kris et al. 2003]. Clinical outcomes were not different between the two doses and the 250 mg/day dose was used for further studies. Based on the results of a placebo-controlled clinical trial of gefitinib in second-line therapy which failed to show an overall survival (OS) benefit in favor of gefitinib, the FDA approval was later withdrawn in 2005 [Thatcher et al. 2005]. Erlotinib, a second EGFR TKI with similar chemical properties, resulted in a 2-month OS benefit over placebo in the second- and third line setting, leading to its FDA approval [Shepherd et al. 2005]. The lessons from this early experience with EGFR-TKI were twofold: (i) responses in the majority of patients were not as great as anticipated, but (ii) major responses that were almost unprecedented with conventional second-line therapies were also observed. Based on clinical parameters, patients with never or only light former smoking status, female gender and particularly those of Asian descent were identified as those who were likely to respond to the EGFR TKI. In addition, the severity of the skin rash which is a classical side effect of EGFR-directed therapies was correlated with better response rates. The identification of activating EGFR mutations in those patients that had a major response to EGFR inhibitors was one of the most fundamental finding in lung cancer biology in recent years [Lynch et al. 2004; Paez et al. 2004], leading to the proof of ‘oncogene addiction’ in a subpopulation of lung cancer patients and invigorating the field of tyrosine kinome sequencing and whole genomic sequencing in lung cancer in an attempt to find additional, novel targets for therapy.
First-line EGFR TKI
EGFR inhibitors have been evaluated in comparison with chemotherapy in previously untreated patients with advanced NSCLC (Table 1).
Overview of recent studies of targeted agents in NSCLC.
The IPASS trial was a multicenter randomized trial performed in several East Asian countries. Patients with advanced pulmonary adenocarcinoma (stage IIIB or stage IV) who had not previously been treated, with no or minimal smoking history (nonsmokers with less than 100 lifetime cigarettes; light former smokers: <10 pack-years and smoking cessation >15 years prior to study entry) were randomized to receive either gefitinib or carboplatin and paclitaxel. The study was designed as a noninferiority study with progression-free survival (PFS) as a primary endpoint [Mok et al. 2009]. The study met its primary endpoint: PFS at 12 months was 24.9% with gefitinib and 6.7% with carboplatin–paclitaxel (hazard ratio [HR] for progression or death: 0.74, p < 0.001). Subgroups were analyzed with respect to the presence of EGFR mutations. Out of 437 evaluable samples, the incidence of EGFR mutations was 261 (59.7%). The vast majority of these were either exon 19 or exon 21 mutations (>95%). It should be noted, however, that about 4% of patients with mutations harbored a T790M mutation, which is associated with primary or acquired resistance to currently used EGFR TKIs. In the subgroup positive for EGFR mutation, PFS was significantly longer with gefitinib than carboplatin–paclitaxel (HR for progression or death, 0.48; p < 0.001); however, in the subgroup negative for the EGFR mutation, the findings were reversed (HR for progression or death, 2.85; p < 0.001). The OS was not significantly different. Carboplatin and paclitaxel was given for a maximum of six cycles. It is unclear how the addition of maintenance therapy or bevacizumab would have influenced the PFS data.
Very similar results were seen in a trial conducted by the North East Japan Study Group. Patients younger than 75 years of age with activating EGFR mutations, but not the T790M mutation were randomized to gefinitib or carboplatin and paclitaxel [Maemondo et al. 2010]. The study was halted prematurely after an interim analysis of 230 patients (of 320 planned patients) revealed that it has met its endpoint of PFS difference. The PFS difference was 10.8 months in the gefitinib arm versus 5.4 months in the carboplatin–paclitaxel arm (HR 0.3, p < 0.001). The median OS difference of 30.5 months versus 23.6 months did not reach statistical significance (p = 0.31).
Similar encouraging results were also seen in another Japanese study, where gefitinib was compared with cisplatin plus docetaxel in patients with NSCLC. This trial only included patients that were positive for the EGFR mutation, as opposed to the IPASS trial which took all comers. In this trial, 177 patients with stage IIIB/IV NSCLC or patients with postoperative recurrence with the EGFR mutation were randomized to receive gefitinib or cisplatin plus docetaxel with an endpoint of PFS. The patients on gefitinib had a significantly longer PFS compared with the cisplatin plus docetaxel group (9.2 months versus 6.3 months, HR 0.489, log-rank p < 0.0001) [Mitsudomi et al. 2010].
The choice of EGFR TKI gefitinib versus erlotinib does not seem to matter much in EGFR-mutated patients. In the OPTIMAL study, 165 patients from China with advanced NSCLC and EGFR mutations were randomized to either erlotinib versus carboplatin and gemcitabine. As expected, patients on erlotinib had significantly superior overall response rates (81% versus 36%, p < 0.0001) and PFS rates (13.6 versus 4.6 months, HR = 0.16, p < 0.0001). OS has not been reached [Zhou et al. 2010].
These results are remarkable, because they represent the first three studies of lung cancer that show a profound PFS benefit for certain patients (EGFR mutated) who are treated with a single targeted agent compared with chemotherapy in the first-line setting. However, almost every patient progressed within 1–2 years of therapy highlighting that resistance to EGFR TKI is a frequent and serous clinical problem.
Earlier attempts to combine both gefitinib (INTACT I and II) [Giaccone et al. 2004; Herbst et al. 2004] and erlotinib (TALENT and TRIBUTE) [Gatzemeier et al. 2007; Herbst et al. 2005] with chemotherapy in the first-line setting were largely without success and may actually demonstrate a trend towards inferior survival in the EGFR TKI plus chemotherapy arms compared with chemotherapy alone. These studies were conducted in unselected patients before predictors for EGFR TKI response were identified. An unplanned subgroup analysis of the TRIBUTE trial demonstrated that never smokers benefited from the addition of erlotinib to chemotherapy [Herbst et al. 2005]. However, it had been unknown whether the addition of chemotherapy to EGFR TKI in EGFR-mutated patients has any benefit. CALGB 30406, a phase II study randomizing never or light former smokers either to erlotinib (E) or erlotinib and chemotherapy with carboplatin and paclitaxel (ECP) found similar response (66% and 69%) and PFS (16.4 versus 17.2 months) rates in both the E and the ECT arm in patients with EGFR mutations. In patients with wild-type EGFR, response rates were 8% (E) and 31% (ECP), favoring chemotherapy as expected [Janne et al. 2010]. A possible explanation for the lack of synergy between EGFR-TKI and chemotherapy may be the induction of G1 cell cycle arrest by EGFR TKIs, leading to increased resistance to chemotherapy [Chang et al. 2004]. A sequential approach using high-dose erlotinib 2 days before chemotherapy has shown promising response and survival rates but awaits confirmation in a phase III trial [Riely et al. 2009].
Second-line EGFR TKI
Erlotinib can be used as second- or third-line treatment in NSCLC, as demonstrated by Shephard and colleagues in the BR.21 trial [Shepherd et al. 2005] (Table 1). Patients who had already failed first- or second-line chemotherapy were randomly assigned to receive either erlotinib or placebo. PFS was 2.2 months in the erlotinib-treated group and 1.8 months in the placebo group. OS was 6.7 months and 4.7 months, respectively (HR 0.70; p < 0.001). At 1 year, 31% of patients treated with erlotinib were alive, whereas 22% of patients on placebo were alive. Predictors of response in multivariate analysis were never smoking status, adenocarcinoma and EGFR expression by immunohistochemistry (IHC) or EGFR amplification by fluorescent in situ hybridization (FISH). Interestingly, EGFR mutations were not a predictor of response in this trial. This finding remains controversial and may be due to the fact that only 197 samples out of 731 patients were analyzed for EGFR mutations and that only 40 of these patients carried an EGFR mutation. Important additional clinical findings of this study are that elderly patients [Wheatley-Price et al. 2008] and those with poor performance status (ECOG PS3) [Shepherd et al. 2005] derive the same clinical benefit as younger, healthier patients and that treatment with erlotinib not only leads to longer survival but also improvement in important quality of life measures [Bezjak et al. 2006].
It remains unclear why the similarly designed ISEL trial comparing gefitinib versus placebo did not show a survival benefit in favor of gefitinib [Thatcher et al. 2005]. In this study, 1692 patients were randomized to either gefitinib or placebo after failure of first-line chemotherapy. Both PFS (3 versus 2.6 months) and OS (5.6 versus 5.0 months) were not statistically significantly different. Subgroup analyses suggest that like in BR.21 EGFR amplification is a predictor for improved survival and that EGFR mutations are a predictor for response [Hirsch et al. 2006]. Furthermore, an unplanned analysis on the subgroup of Asian patients revealed a significant PFS for this population (4.4 versus 2.2 months, p = 0.084) [Chang et al. 2006]. It had been postulated that one possible reason for gefitinib’s failure was the fact that unlike erlotinib in the BR.21 study, it was dosed significantly below its maximum tolerated dose (MTD). However, the early phase II trials IDEAL I and II in which equivalent responses and survivals were observed between the 250 and 500 mg per day doses seem to contradict this hypothesis [Fukuoka et al. 2003; Kris et al. 2003]. Further complicating the assessment of gefitinib in the second line setting are the results of the INTEREST study [Kim et al. 2008]. Here, 1466 patients were randomized to either gefitinib 250 mg per day or docetaxel, a standard second-line regimen for NSCLC. Gefitinib achieved its primary endpoint of noninferiority in regards to OS, possibly establishing it as a valid second-line treatment strategy for NSCLC. A biomarker analysis was performed on 453 evaluable samples. EGFR mutation status, EGFR copy number, and EGFR protein expression were not correlated with improved OS from gefitinib. However, EGFR mutations were associated with higher objective response rate (41.1% versus 21.1%, p = 0.04) and improved PFS (HR 0.16, p = 0.001). EGFR amplification by FISH was associated with a higher objective response rate (13.4% versus 7.1%, p = 0.4) [Douillard et al. 2010].
Maintenance therapy
Maintenance therapy or early second-line therapy is a relatively new concept in the treatment of NSCLC, gaining mainstream acceptance last year after the presentation of a seminal trial which showed dramatically improved OS rates in NSCLC with nonsquamous histology when treated with maintenance pemetrexed [Ciuleanu et al. 2009]. Other maintenance options based on ECOG 4599 and the FLEX trial are bevacizumab and cetuximab [Pirker et al. 2009; Sandler et al. 2006]. The SATURN trial evaluated erlotinib in this setting (Table 1); of the 1949 patients initially included, 889 patients without disease progression were randomly assigned to receive erlotinib or placebo daily. Median PFS was longer in patients receiving erlotinib (12.3 weeks versus 11.1 weeks; HR 0.71; p < 0.0001), in all patients, as well as in the subgroup that was EGFR receptor positive. In patients that received erlotinib, 25% had not progressed at 6 months, as opposed to 15% in the placebo group. Complete or partial response occurred in 11.9% of the erlotinib group versus 5.4% of the placebo group (p = 0.0006). OS also was improved with erlotinib, 12.0 months versus 11.0 months (HR 0.81; p = 0.0088). While the benefit from erlotinib overall appears modest, the subgroup of patients with EGFR mutations not unexpectedly experienced a substantial prolongation on PFS (HR 0.1, p < 0.001) [Cappuzzo et al. 2010].
Cetuximab
Although several trials showed that the addition of erlotinib and gefitinib to first-line chemotherapy did not improve outcome in patients with advanced NSCLC, two studies examining the role of the EGFR monoclonal antibody cetuximab with chemotherapy were conducted recently: the phase III FLEX trial compared cetuximab in combination with cisplatin and vinorelbine versus cisplatin and vinorelbine alone [Pirker et al. 2009] in 1125 patients. Cetuximab was continued until major toxicity or progression of disease. OS was significantly longer in the group receiving cetuximab plus chemotherapy as compared with chemotherapy alone, 11.3 months versus 10.1 months (HR for death: 0.871, p = 0.044). At 1 year, 47% of the cetuximab group was alive as compared with 42% in the chemotherapy alone group. Although OS was improved favoring cetuximab, PFS was not. The second slightly smaller study with 676 patients (BMS099) compared cetuximab with carboplatin and paclitaxel versus chemotherapy by itself [Lynch et al. 2010]. The trial failed to reach its primary endpoint of an improvement in PFS (4.4 versus 4.24 months, HR 0.9, p = 0.236). While there was a trend towards improved OS, those results were not statistically significant (9.69 months versus 8.38 months with TC; HR 0.890; p = 0.169). Interestingly, k-ras mutation status, a strong predictor against cetuximab response in colorectal cancer [Van Cutsem et al. 2009], did not predict against cetuximab response in either trial [Khambata-Ford et al. 2010].
EGFR inhibitors in the adjuvant setting
Results of a randomized phase III study (NCIC BR19) comparing gefitinib versus placebo for 2 years in patients with resected stage IB to IIIA NSCLC have been presented recently [Goss et al. 2010]. The study was closed prematurely in 2005 after the results of the ISEL study cast doubt on the efficacy of gefitinib in unselected patients. Overall 503 patients were enrolled. Provocatively, there was a trend towards both inferior PFS (HR 1.22, p = 0.15) and OS (HR 1.24, p = 0.14). K-ras mutation (available in 350 patients) and EGFR copy number (available in 348 patients) were not predictive of response. While a reason for a possible detriment from EGFR TKI treatment is not immediately clear, these data raise concern since gefitinib consolidation therapy has been associated with a trend towards inferior OS once before, although in a different setting: in the SWOG S0023 study, maintenance gefitinib after consolidation docetaxel following concurrent chemotherapy with thoracic radiotherapy in patients with stage III NSCLC, the median OS was 23 months with gefitinib versus 35 months with placebo [Kelly et al. 2008]. In the still accruing randomized RADIANT trial evaluating the role of erlotinib, patients whose tumors are positive by EGFR IHC are eligible for participation. Other studies allow only patients with EGFR mutated tumors to participate.
Resistance in EGFR-mutated patients
Even in EGFR-mutated patients resistance to EGFR inhibitors usually occurs within 1 year. Therapies to target possible resistance mechanisms are currently either in preclinical or early stage clinical development. Irreversible EGFR inhibitors have activity in tumors with a T790M mutation. However, these are not specific for T790M and inhibition of wild-type EGFR occurs as well, possibly leading to increased toxicity. In early clinical trials in unselected pretreated patients response rates of irreversible EGFR inhibitors were low [Janne et al. 2007], even in patients with known EGFR mutations [Sequist et al. 2010]. A potentially promising new approach is the recent development of specific inhibitors against EGFR T790M which do not inhibit wild-type EGFR [Zhou et al. 2009]. However, clinical data on these compounds do not exist to date. Phase I/II clinical trials are currently underway to study the potential synergy between EGFR and met inhibition (PF-00299804 ± crizotinib [ClinicalTrials.gov identifier: NCT00965731]) in patients with acquired resistance against erlotinib or gefitinib and synergy between erlotinib and the IGFR TKI OSI-906 as first-line treatment in patients with EGFR mutations [ClinicalTrials.gov identifier: NCT01221077].
VEGF
Since all tumors are dependent on neo-angiogenesis, it is not surprising that VEGF directed therapy was among the first targeted therapy to be studied for NSCLC (Table 1). Monoclonal antibodies (bevacizumab), recombinant VEGF receptors (aflibercept), and small-molecule VEGFR inhibitors (sorafenib, vandetinib) are available agents.
A phase II trial comparing bevacizumab in combination with carboplatin and paclitaxel versus carboplatin and paclitaxel alone showed an improvement in time to progression and OS (7.4 versus 4.2 months, and 17.7 versus 14.9 months, respectively) [Johnson et al. 2004]. However, a large number of life-threatening and often fatal hemoptyses were observed, most of them in patients with squamous cell carcinomas, which often cavitated. As a consequence, patients with squamous histology were excluded from further trials with bevacizumab.
The subsequent phase III trial (E4599) compared chemotherapy with paclitaxel–carboplatin alone or with bevacizumab in NSCLC [Sandler et al. 2006]. A total of 878 patients with recurrent or advanced NSCLC were treated with either paclitaxel–carboplatin alone or in combination with bevacizumab. Bevacizumab was continued until disease progression. OS was longer in the group that received bevacizumab with chemotherapy, 12.3 months versus 10.3 months (p = 0.003). One- and two-year survival rates were 51% and 23% versus 44% and 15% for the chemotherapy plus bevacizumab group versus chemotherapy alone. PFS was also longer in the paclitaxel– carboplatin–bevacizumab group, 6.2 months versus 4.5 months (p < 0.001).
In the AVAiL trial, bevacizumab (at 7.5 mg/kg and 15 mg/kg) in combination with cisplatin and gemcitabine was compared versus placebo in 1043 patients. Patients receiving bevacizumab 7.5 mg/kg derived the most benefit, with an improvement in PFS compared with placebo (6.7 versus 6.1 months, HR 0.71, p = 0.023) [Reck et al. 2009]. PFS surprisingly was similar in the group receiving 15 mg/kg of bevacizumab as compared with placebo (6.5 versus 6.1 months, HR 0.84; p = 0.25). OS was not significantly different between the three groups (13.6 (B 7.5 mg) and 13.4 months (B 15 mg) versus 13.2 months (placebo), p = NS) [Reck et al. 2010]. Notably, the group receiving the higher dose of bevacizumab actually did not do better than the lower dose. Patients with brain metastases were excluded from both trials due to concerns that bevacizumab may increase the risk of intracranial hemorrhage. The safety of bevacizumab in patients with treated brain metastases was subsequently established by the PASSPORT trial [Socinski et al. 2009] and generally manageable toxicity of bevacizumab was confirmed in the SAIL study, an open-label phase IV study, which did not reveal any new safety signals [Crino et al. 2010].
The experience with small-molecule inhibitors of VEGFR2 has been largely disappointing. Sorafenib is a TKI that in addition to VEGFR2 targets c-kit, raf, and PDGF. The randomized ESCAPE trial evaluating the role of sorafenib in combination with first-line chemotherapy in NSCLC failed to show any improvements in either OS or PFS when compared with the chemotherapy alone arm [Scagliotti et al. 2010]. Results with vandetanib, a VEGFR, EGFR, and RET antagonist have been equally disappointing. While the second-line ZODIAC study in combination with docetaxel showed a PFS advantage favoring the addition of vandetanib to chemotherapy over chemotherapy alone (PFS 4 versus 3.2 months, p < 0.001), there was no difference in OS [Herbst et al. 2010]. In combination with pemetrexed vandetanib failed to show improved PFS or OFS when compared with pemetrexed alone (ZEAL trial) [De Boer et al. 2009]. Finally, vandetanib also failed to prove superior over erlotinib in patients in the second-line setting (ZEST trial) [Natale et al. 2009]. As third-line therapy after progression to erlotinib, vandetanib was not superior to placebo [Lee et al. 2010]. Currently sorafenib is undergoing testing as fourth-line agent in NSCLC against placebo and best supportive care (BSC).
Should the ‘angiogenic switch’ hypothesis be true, one would expect perhaps the most significant benefit from anti-angiogenic therapy in the postoperative setting where the development of new metastases has to depend on new blood vessel formation. This question is currently being addressed in the ECOG1505 study, which randomizes patients with stage IB–IIIA NSCLC to four cycles of cisplatin-based chemotherapy ± bevacizumab for 1 year. The fact, however, that two adjuvant studies in colorectal cancer (NSABP-C08 and AVANT) failed to show a benefit from adjuvant therapy with bevacizumab for 1 year in combination with chemotherapy may diminish the enthusiasm for this approach.
Combined EGFR and VEGF blockade
Since both VEGF and EGFR pathways play critical roles in the biology of NSCLC, an approach aimed at those two targets seems reasonable. Phase II studies using the combination of erlotinib, bevacizumab versus chemotherapy plus bevacizumab versus chemotherapy alone were promising [Herbst et al. 2007]. The subsequent and so far unpublished randomized phase III study comparing erlotinib ± bevacizumab (BeTa trial) showed a significant improvement in both response rates (12.6% versus 6.2%) and PFS (3.4 versus 1.7 months, p < 0.001), but failed to meet its primary endpoint of showing an OS benefit of the combination therapy (9.3 versus 9.2 months, p = NS). The combination of cetuximab, avastin, and chemotherapy has also shown very promising response rates in first-line phase II trials [Gandara et al. 2010], but we are awaiting confirmation of those results in randomized fashion, particularly in light of recent evidence in colorectal cancer that the combination may actually shorten PFS [Tol et al. 2009].
Anaplastic lymphoma kinase
Recent evidence has shown that a small subset of adenocarcinomas carry the EML4-Alk fusion protein, leading to overexpression of Alk in these cancers. These cancers may be ‘oncogene addicted’ since targeted overexpression of the EML4-Alk protein in type II pneumocytes is sufficient to cause lung cancer in mice [Chen et al. 2010]. Evaluation of the dual Alk and Met inhibitor crizotinib in a phase II study of patients with Alk translocation created much excitement because the observed results mimicked those outcomes in patients with EGFR mutations who were treated with EGFR inhibitors [Kwak et al. 2010]. Overall response rates were 57% (1CR, 46PR), the disease control rates 90%. The median survival rates have not been reached in this study. None of the tumors tested was positive for Met amplification leading to the conclusion that crizotinib’s function as a Met inhibitor did not contribute to those results. The molecular basis of resistance in the 10% of patients with progressive disease is not yet clear. In a cell line model, however, co-activation of Her-2 and EGFR has been described as a potential mechanism of resistance [Koivunen et al. 2008].
Epigenetic therapy
Both DNMTs and HDACs play are crucial role in the silencing of tumor suppressor genes and in the development of lung cancer. HDACs also modify numerous critical nonhistone proteins such as p53, beta-catenin and alpha-tubulin. DNMT inhibitors and HDAC inhibitors are currently in early clinical development for NSCLC. In a randomized phase II study the HDAC inhibitor vorinostat in combination with chemotherapy (carboplatin and paclitaxel) led to very encouraging results in regard to response rates when compared with chemotherapy alone (risk ratio [RR] 34% versus 12%, p = 0.02) [Ramalingam et al. 2010b]. The study was not powered to detect a survival benefit, but trends favoring both PFS (6.0 versus 4.1 months, p = 0.48) and OS (13.0 versus 9.7 months, p = 0.17) were observed.
PI3K–Akt–mTOR
The mTOR pathway is one of the most commonly activated pathways in cancer. The mTOR inhibitors temsirolimus and everolimus are FDA approved for the treatment of renal cell carcinoma, where they have significant single-agent activity. For lung cancer, they are still in early clinical development. At our institution, we have recently completed a phase I trial of everolimus together with docetaxel in the second-line treatment for NSCLC [Ramalingam et al. 2010a]. Tumor responses were promising. PI3K inhibitors are currently in phase I clinical evaluation together with chemotherapy (GDC-0941 [ClinicalTrials.gov identifier: NCT00974584]) or erlotinib (XL765 [ClinicalTrials.gov identifier: NCT00777699] or XL147 [ClinicalTrials.gov identifier: NCT00692640]). The Akt inhibitor (MK-2206) is in development as a single agent, together with chemotherapy and in combination with gefitinib in phase I studies.
IGFR
IGFR is a potent activator of the PI3K-Akt-mTOR pathway and has also been implicated in resistance to EGFR inhibitors through heterodimerization of the receptor with EGFR. Since it is frequently overexpressed in NSCLC it is an attractive potential therapeutic target. A randomized phase II study comparing chemotherapy ± the monoclonal antibody against IGFR, figitumumab, was encouraging (RR 54% versus 42%) [Karp et al. 2009]. The study met the primary endpoint of rejecting the null hypothesis of a RR of 28% with chemotherapy alone, although response rates in the chemotherapy only arm were also impressive. However, a subsequent randomized phase III study randomizing patients to carboplatin/paclitaxel ± figitumumab closed prematurely with 681 out of 820 patients enrolled after a preplanned interim analysis showed that the trial would not meet its primary endpoint of an OS difference) [Jassem et al. 2010]. Most patients on this study (86%) had squamous cell carcinomas. A second monoclonal antibody IMC-A12 is currently under investigation in various combinations (chemotherapy alone, chemotherapy with bevacizumab, chemotherapy with cetuximab, erlotinib) in NSCLC. Small-molecule inhibitors against IGFR are in early phase clinical trials.
Conclusion
Targeted therapies have revolutionized both the treatment of NSCLC as well as our understanding of the underlying molecular pathways. The magnitude of negative studies highlights the fact these treatments are not ‘one size fits all’. In this light, a negative study may be more a reflection of an unselected patient population rather than disproof of a certain principle. It is of critical importance even in negative trials to identify molecular signatures that are predictive of response and to have information flow from bench to bedside and back. The identification of the EGFR mutations is so far the best example of a successful strategy in which clinical observation dramatically influenced the laboratory science. We are hopeful that similar results can be achieved at least for some of the many other targets that are currently being studied. The future will hold dramatically new approaches to lung cancer therapy, but will also pose new challenges to oncologists, pulmonologists, and pathologists alike. The term NSCLC will likely disappear as tumors will likely be classified based on their molecular signatures, which are likely to change with the emergence of resistance to any particular therapy. The requirement for more frequent and larger biopsies, and novel methods of molecular diagnosis will likely grow as the complexities of the underlying treatments do.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
None declared.