
Abstract
Genetic variations in the host TLRs genes play an important role in susceptibility and/or resistance to visceral leishmaniasis by altering the host-pathogen interaction. In this study, we investigated the association between polymorphisms of TLR4 (Asp299Gly, Thr399Ile) and TLR-9 (T-1237C), with susceptibility to visceral leishmaniasis. A bi-directional PCR amplification of specific alleles technique was used to characterize the distribution of TLR4 (Asp299Gly and Thr399Ile) and TLR9 (T-1237C) polymorphisms. A total of 60 samples were randomly selected from confirmed visceral leishmaniasis patients and 24 endemic healthy volunteers. The samples were genotyped and allele frequencies were determined. We observed that TLR4 Asp299Gly and Thr399Ile genotypes were more frequent in visceral leishmaniasis patients (10% and 15% respectively) compared to controls (4.2% and 8.3% respectively). However, the differences were not significant in TLR4 Asp299Gly and Thr399Ile alleles and genotypes. In the case of TLR9, we observed the frequency of T1237C genotype was higher in visceral leishmaniasis patients (43.3%) than in healthy controls (33.3%). Statistically significant differences were observed in TLR9 T1237C alleles and genotypes. We concluded that TLR9 T1237C, but not TLR4, gene polymorphisms can be regarded as contributors to visceral leishmaniasis susceptibility among the Indian population of Bihar state.
Introduction
Leishmaniasis is a spectrum of diseases caused by the protozoan parasite Leishmania sp. and is transmitted to humans and other mammals by the bite of female phlebotomine sandflies. 1 , 2 Leishmaniasis is endemic in 97 countries and territories and four countries have previously reported cases. More than 616 million people from endemic areas are at a risk of infection. 3 The global burden of leishmaniasis is 300,000 cases annually with over 20,000 deaths per year. Among the different forms of leishmaniasis, visceral leishmaniasis (VL, or kala-azar) is the most severe and is fatal if left untreated. In 2015, almost 90% of global VL cases were reported from six countries: India, Brazil, Ethiopia, Somalia, Sudan and South Sudan. In India, VL is presently endemic in four states comprising 54 districts in the country; of these 33 districts are from Bihar state, which contributes to >70% of total VL cases reported from India. 3 Recent studies demonstrated the immune status. 4 Investigation of the effects of susceptible genes on VL will provide additional information about the molecular genetic mechanisms involved in leishmaniasis development and aid in the establishment of more efficient ways to prevent the spread of leishmaniasis.
TLRs are a class of PRR proteins expressed on cells of the innate immune system and are crucial for recognition of motifs on pathogen surfaces (PAMPs), thus triggering protective inflammatory responses. The interaction of pathogens with different TLRs contributes to the outcome of infection. 5 TLR4 specifically recognizes LPS motifs on the surfaces of the invading pathogen whereas TLR9 preferentially binds to unmethylated CpG sequences in DNA molecules and triggers a series of signaling events that lead to expression of pro-inflammatory genes such as cytokines, which might result in symptomatic disease. 5
Previously, we have shown the importance of TLR4 and TLR9 signaling in VL. We have shown that TGF-β1-mediated TLR4 signaling promotes L. donovani infection through enhancement of SHP-1 and ubiquitin-edited enzyme A20. 6 Further, we have shown that combination of paromomycin and miltefosine, a model antileishmanial therapy for VL, targets the host TLR functions. 7 Our results revealed that paromomycin and miltefosine interact with TLR4 and lead to NF-kB promoter activation through MyD88. Either as monotherapy or in combination, these drugs kill macrophage-bound L. donovani by inducing release of TNF-α and NO in a TLR4-dependent manner. 7 In another study, we showed that paromomycin/miltefosine have a TLR9-mediated synergistic effect on host immune responses and it renders protection against Leishmania infection via TLR9. 8 We observed that paromomycin/miltefosine interact with TLR9 and up-regulate NF-κB promoter activity through MyD88, leading to increased stimulation of naive T cells to produce IFN-γ. 8 Apart from TLR4, we have also explored the involvement of TLR9 in the disease outcome of VL. We observed that the L. donovani genome containing species-specific CpG motifs interacts with TLR9 and plays an important role in activating the innate immune system and regulation of macrophage-programmed cell death and parasite survival inside macrophages. 9
Polymorphism in the genes of TLRs results in abnormal host-pathogen interaction due to variation in TLR proteins and thus, anomalous immune response. Several single-nucleotide polymorphisms (SNPs) have been identified for TLR4, and TLR9, altering susceptibility to infectious and inflammatory diseases. 10 For TLR4, two major polymorphisms (Asp299Gly and Thr399Ile) were observed to reduce reactivity to inhaled LPS, 11 , 12 although findings are partly conflicting. 10 , 13 Individuals exhibiting Asp299Gly and Thr399Ile SNPs are at increased risk of septic shock 14 and Gram-negative infection, 15 respectively. Moreover, other rare TLR4 mutations have been described to increase susceptibility to meningococcal meningitis. 16 , 17 Two common TLR9 promoter polymorphisms (T-1237C and T-1486C), assumed to influence transcription regulation, have been described in African Americans, one of them (T-1237C) being potentially associated with asthma 18 and Crohn’s disease. 19
There are very few studies on the association between TLR polymorphisms and susceptibility to VL. To our knowledge, there is no previous study on the association between polymorphisms of TLRs (TLR4 and TLR9) and susceptibility to VL caused by L. donovani from India. In the present study, we examine the role of TLR4 (Asp299Gly, Thr399Ile) and TLR-9 (T-1237C) polymorphisms in susceptibility to VL in the Indian population in Bihar state.
Materials and methods
Study subjects
In this case-control study, we randomly recruited a total of 60 VL patients (≥ 5 to ≤ 60 yr age group) admitted to the indoor ward of Rajendra Memorial Research Institute of Medical Sciences. VL was diagnosed by clinical symptoms, serological and parasitological tests following the standardized protocol. An additional 24 healthy individuals (who were volunteers) (≥ 5 to ≤ 60 yr age group) living in the same rural endemic areas acted as an endemic control and were included in this study, with both males and females included. Every fifth patient admitted to the indoor ward of Indian Council of Medical Research (ICMR) Rajendra Memorial Research Institute of Medical sciences was randomly recruited in the study. Randomization was implemented to stop the introduction of bias. The health or disease status of the individuals was checked and confirmed by a medical doctor before recruitment to the study. Healthy individuals were confirmed after confirmation of no previous report of VL/Post Kala-azar Dermal Leishmaniasis (PKDL), rK39 negative and no present illness. Written informed consent was obtained from adult individuals. Moreover, informed consent was obtained from legal guardians of the children. Approval for the study was provided by the Institutional Ethical Committee of Rajendra Memorial Research Institute of Medical Sciences. VL patients co-infected with HIV or other cross-reactive diseases including tuberculosis, malaria, vitiligo and so on were excluded from the study.
DNA isolation
Blood samples (∼1 ml) were collected in lithium-heparin vacutainers (BD Biosciences, San Jose, CA, USA) after written informed consent was received from the volunteers. Genomic DNA was isolated from peripheral blood by a Qiagen DNA Blood Minikit (Qiagen, Hilden, Germany) as per the manufacturer’s protocol.
TLR4 and TLR9 genotyping
The Bi-PASA method was developed to distinguish between homozygotes and heterozygotes in one PCR reaction. 20 , 21 Briefly, this method allows two allele-specific amplifications occurring in opposite directions, with the mismatches at the 3’ end of each primer. It is well adapted for studies attempting to determine whether there is an association between a SNP in a gene and a pathological phenotype in a human cohort. Bi-PASA is performed with four primers: two outer primers, P and Q, and two inner allele-specific primers, M and W (Tables 1 and 2). P and Q anneal at different distances from the sequence change to differentiate the downstream and upstream PASA assays on an agarose gel. M and W are each specific to an allele with the mismatch at (or near) the 3’ end of the primer. Depending on the zygosity, Bi-PASA produces two or three overlapping fragments. PQ is always produced and serves as a positive control. PW and MQ are both present in a heterozygote (WT/M), but only PW is produced in homozygous wild type (WT/WT) and only MQ is produced in homozygous mutant (M/M) samples (Table 1). Primers for Bi-PASA were designed following the guidelines proposed by Liu et al. and Carvalho et al., 20 , 21 taking into account both the melting temperature of both the primers and the largest PCR segment (PQ). The primers used in this study are shown in Table 2.
Diagrammatic representation of display of polymorphism on agarose gel.

Primers used for the Bi-PASA method.

Bi-PASA: bi-directional PCR amplification of specific alleles.
The Bi-PASA methodology was applied using DNA samples from individuals. DNA was isolated from whole blood samples using a Qiagen DNA Blood Minikit (Qiagen, Hilden, Germany) and PCR amplification was performed in a 25 µl volume that included nuclease free water, PCR buffer (10×), deoxyribonucleotide triphosphate (dNTP) mixture (2.5 mM each), primers (0.05–0.4 mM each; for details see Table 2), Taq DNA polymerase (1.25 U/25 µl) and approximately 50 ng genomic DNA templates.
Primers were obtained from Integrated DNA Technologies (IDT, Coralville, Iowa, USA) and other PCR reagents were purchased from Takara (New Delhi, India). PCR cycling conditions included 35 cycles of 15 s at 94°C, 45 s at 57°C and 45 s at 67°C, after a 10 min initial period of DNA denaturation and enzyme activation at 94°C. The amplified fragments had sizes readily distinguishable by electrophoresis in a 1.5% agarose gel (USB, Cleveland, Ohio, USA). The images of gels were captured by a Chemidoc Gel imaging system using Quantity One software (Bio-Rad, Hercules, California, USA).
Statistical analysis
Genotype distribution and comparison of the polymorphisms of the patient and control groups was tested for Hardy-Weinberg equilibrium by Chi-square (χ2) analysis.
Results and discussion
TLR4 and TLR9 gene polymorphisms were analysed in all healthy and VL in three groups, namely (a) TLR4 (Asp299Gly), (b) TLR4 (Thr399Ile) and (c) TLR9 (T1237C). The genotype and allele frequencies of the gene polymorphisms among cases and controls are shown in Tables 3--5. In the present study, we investigated the association between TLR4 (Asp299Gly and Thr399Ile) and TLR 9 (T1237C) gene polymorphisms to susceptibility of VL among population from endemic districts of Bihar, India. The band pattern for TLR4 and TLR9 obtained after PCR amplification of VL and endemic healthy controls are shown in Figures 1–3.
Summary of results obtained.
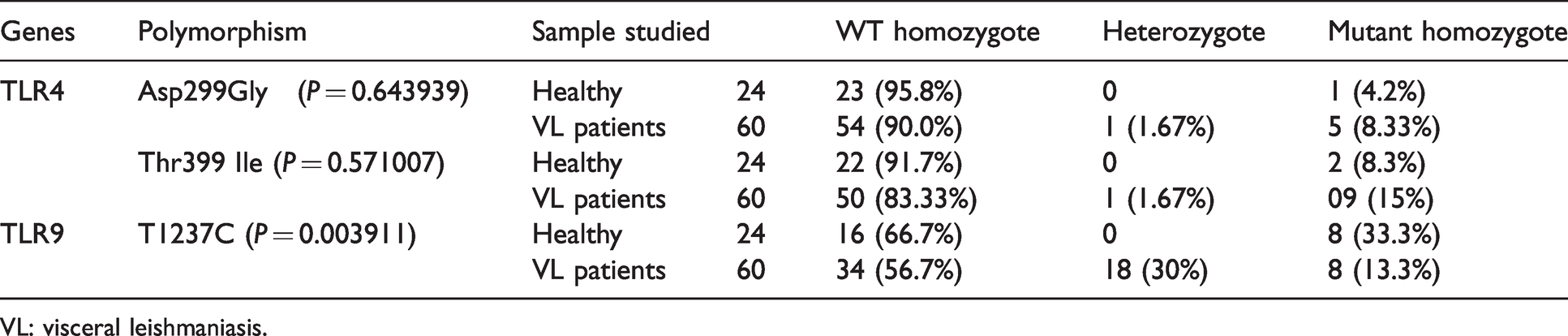
VL: visceral leishmaniasis.
Allele and genotype frequencies of TLR4 and TLR9 gene variants in healthy groups and VL patients.
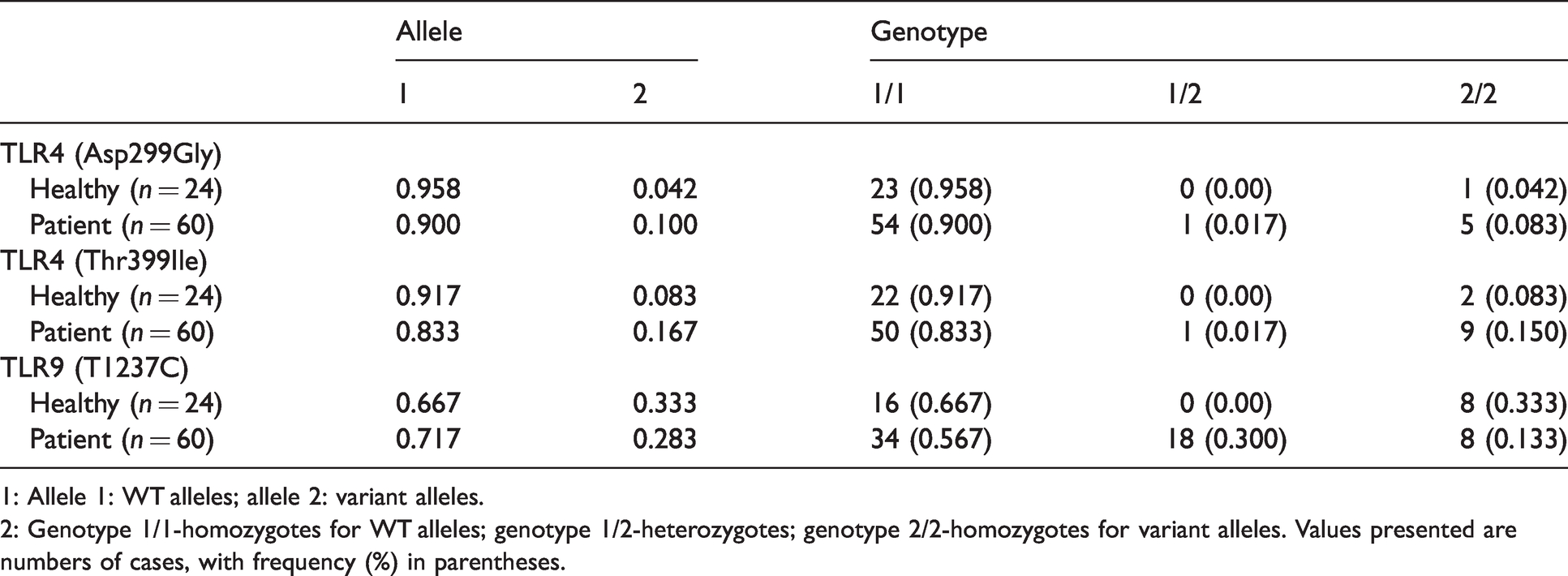
1: Allele 1: WT alleles; allele 2: variant alleles.
2: Genotype 1/1-homozygotes for WT alleles; genotype 1/2-heterozygotes; genotype 2/2-homozygotes for variant alleles. Values presented are numbers of cases, with frequency (%) in parentheses.
Genotype frequencies of TLR4 and TLR9 gene variants in patient and healthy control.
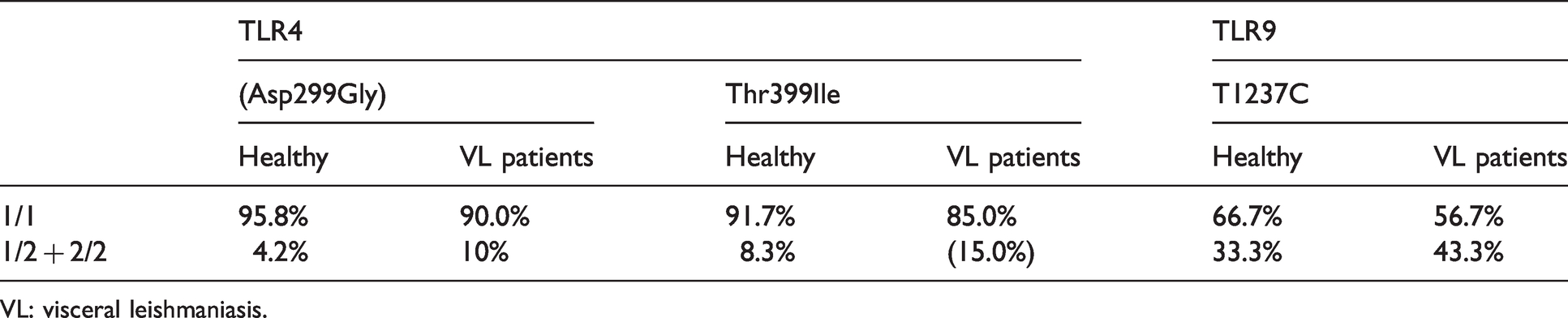
VL: visceral leishmaniasis.
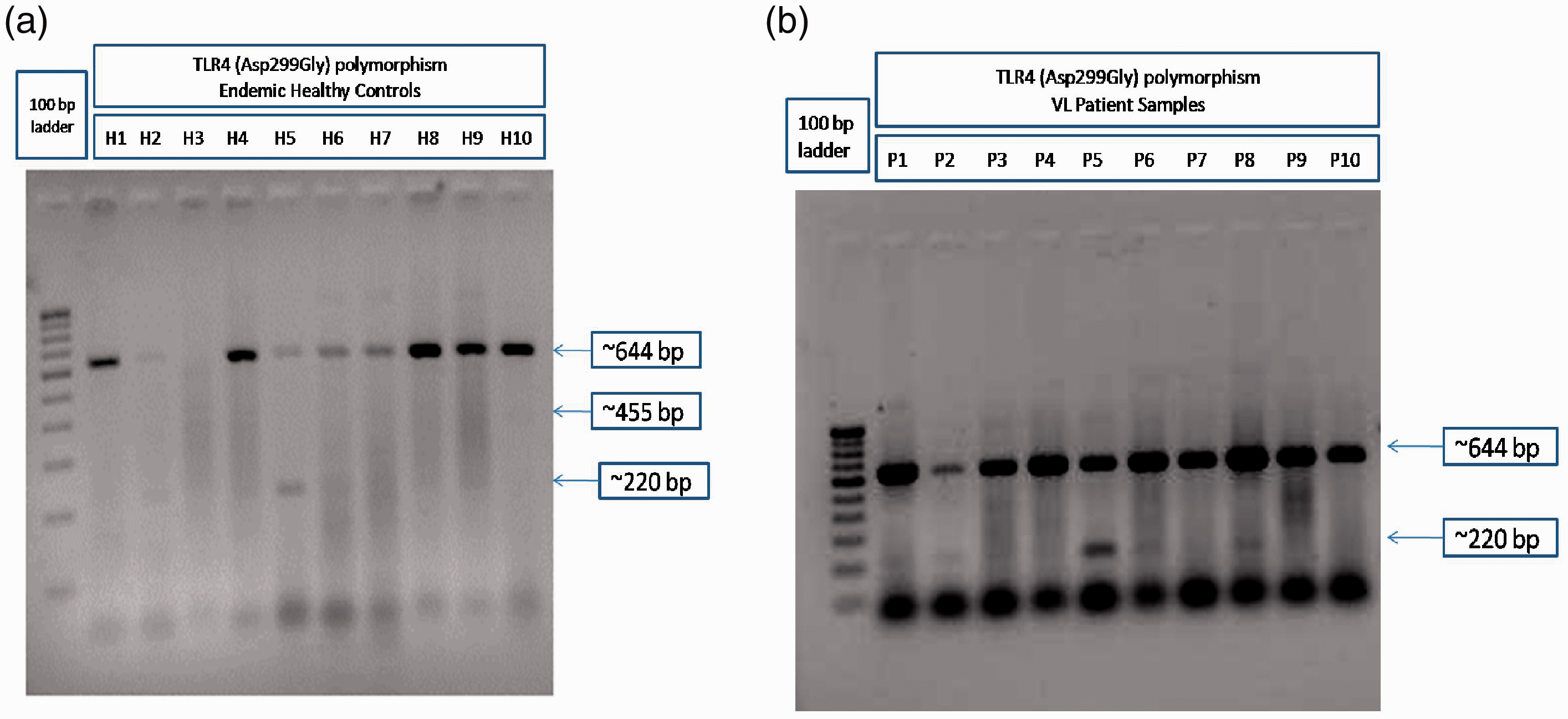
Representative gel image of bi-directional PCR amplification of specific alleles (Bi-PASA) genotyping for TLR4 (Asp299Gly) of endemic healthy control and visceral leishmaniasis (VL) patient samples. Endemic healthy control (a; H1-H10) and VL patient (b; P1-P10) DNA samples were genotyped for TLR4 (Asp299Gly) polymorphism by the Bi-PASA method using gene-specific primers. The PCR products were run on 1.5% agarose gel and photographed in a gel documentation system (Bio-Rad) and one of the representative gel images was provided.
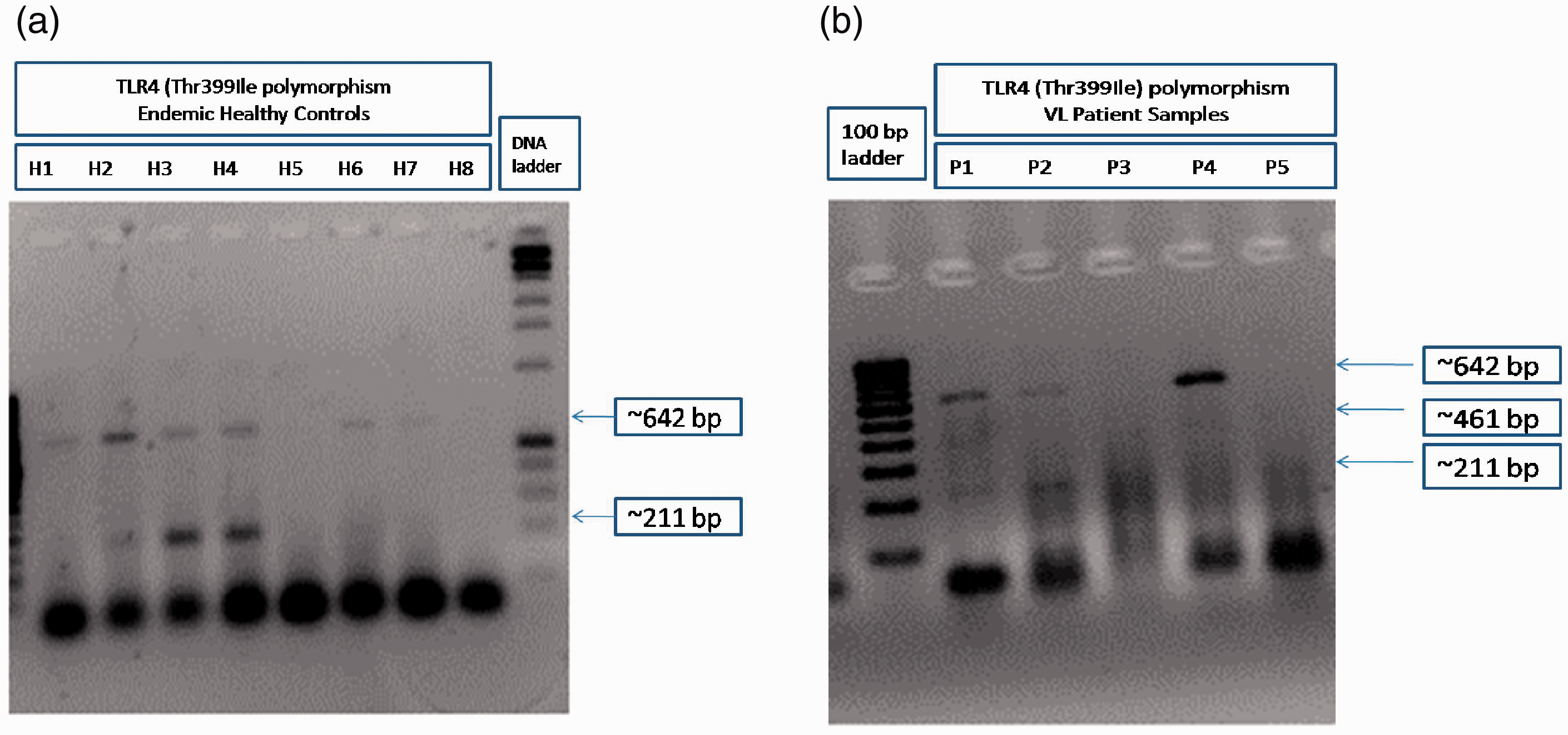
Representative gel image of bi-directional PCR amplification of specific alleles (Bi-PASA) genotyping for TLR4 (Thr399Ile) of endemic healthy control and visceral leishmaniasis (VL) patient samples. Endemic healthy control (a; H1-H8) and VL patient (b; P1-P5) DNA samples were genotyped for TLR4 (Thr399Ile) polymorphism by the Bi-PASA method using gene-specific primers. The PCR products were run on 1.5% agarose gel and photographed in a gel documentation system (Bio-Rad) and one of the representative gel images was provided.
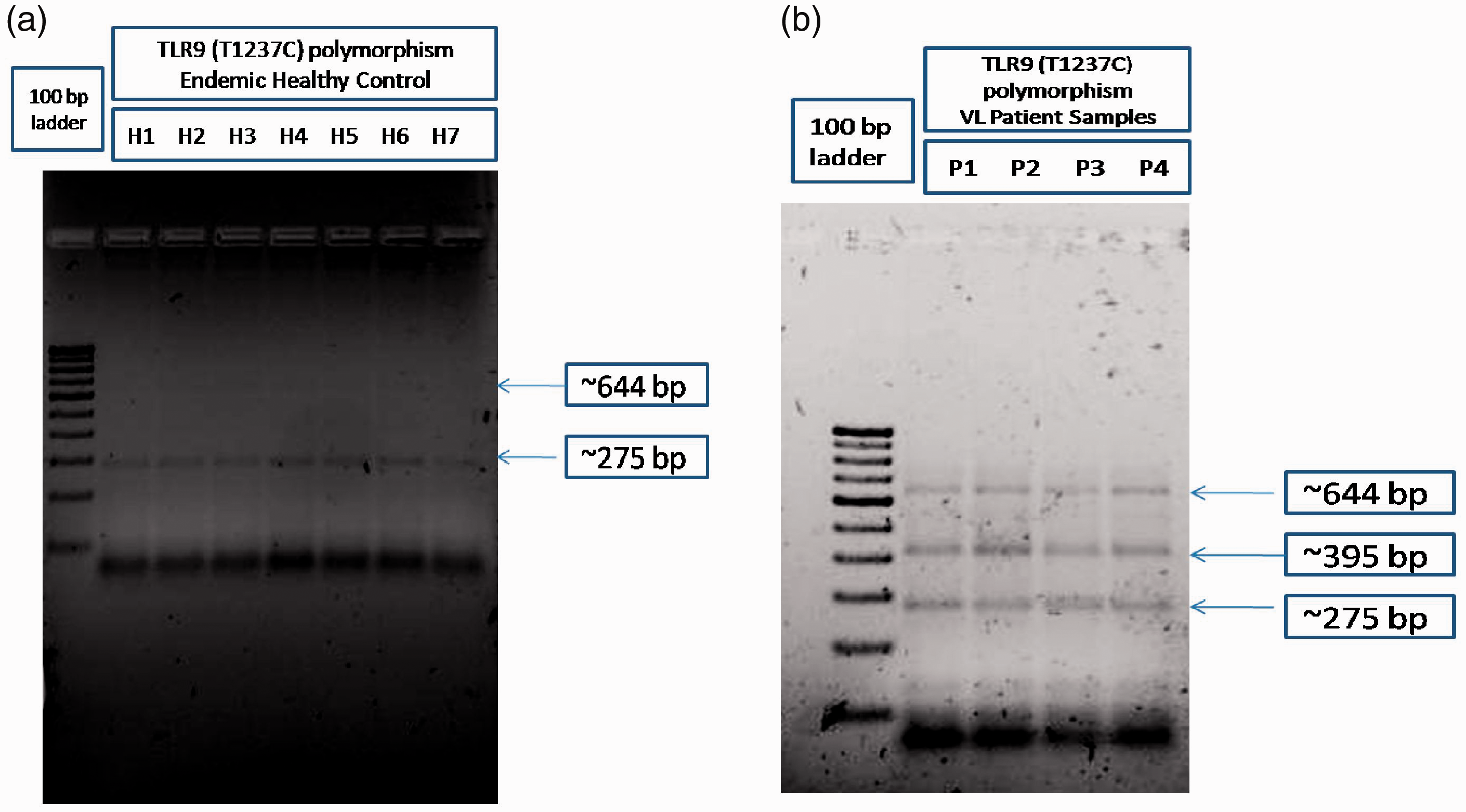
Representative gel image of bi-directional PCR amplification of specific alleles (Bi-PASA) genotyping for TLR9 (T1237C) of endemic healthy control and visceral leishmaniasis (VL) patient samples. Endemic healthy control (a; H1-H7) and VL patient (B; P1-P4) DNA samples were genotyped for TLR9 (T1237C) polymorphism by the Bi-PASA method using gene specific primers. The PCR products were run on 1.5% agarose gel and photographed in a gel documentation system (Bio-Rad) and one of the representative gel images was provided.
After obtaining written informed consent from each individual, a total of 60 randomly selected confirmed VL patients and 24 healthy volunteers were genotyped using Bi-PASA. Among the tested individuals, 23/24 (95.8%) and 54/60 (90.0%) were WT homozygous; 01/24 (4.2%) and 05/60 (8.33%) were mutant homozygous for Asp299Gly TLR4 polymorphisms in healthy and VL patients, respectively (Table 3). In contrast, none of the healthy individuals were heterozygous whereas 1/60 (1.67%) was heterozygous in VL patients for Asp299Gly TLR4 polymorphism (Table 3).
Moreover, in the case of TLR4 Thr399Ile polymorphism, 22/24 (91.7%) and 50/60 (83.33%) were found to be WT homozygous; 2/24 (8.3%) and 9/60 (15%) were found to be mutant homozygous in healthy and VL patients respectively (Table 3). None of the healthy individuals 1/60 (1.67%) VL patients were heterozygous for Asp299Gly TLR4 polymorphism (Table 3).
However, in the case of T1237C TLR9 polymorphisms, 16/24 (66.7%) and 34/60 (56.7%) were WT homozygous; 8/24 (33.3%) and 8/60 (13.3%) were mutant homozygous in healthy and VL patients respectively among the tested individuals (Table 3). No healthy individuals were heterozygous for T1237C TLR9 polymorphisms. However, 18/60 (30.0%) VL patients were heterozygous for T1237C TLR9 polymorphism (Table 3).
The distribution of these genotypic variants met the Hardy-Weinberg equilibrium. The results showed the Asp299Gly genotype was more frequent in patients (10%) than in healthy controls (4.2%). However, the difference was not statistically significant (P = 0.643939) (Tables 3--5). Moreover, it was observed that the frequency of Thr399Ile genotype was higher in the VL patients (15%) than in the controls (8.3%). However, the difference was not statistically significant (P = 0.571007) (Tables 3--5).
Our results are in concordance with the previous studies on other parasitic infections where it was observed that the frequencies of TLR4 Asp299Gly and Thr399Ile polymorphisms did not show any significant differences in the distribution of alleles or genotypes between patients with Chagasic cardiomyopathy and asymptomatic subjects. 22 However, in another study on neurocysticercosis (NCC), it was observed that TLR4 Asp299Gly and Thr399Ile polymorphisms were associated with its occurrence (P < 0.001 for Asp299Gly and P = 0.003 for Thr399Ile) and progression to symptomatic NCC, compared with control subjects or asymptomatic NCC. 23
In the case of leishmaniasis, Ajdary et al. also studied the relationship between TLR4 polymorphisms and cutaneous leishmaniasis (CL). They investigated the role of TLR4 mutants (Asp299Gly and Thr399Ile) in the outcome of CL. They reported that AG (Asp299Gly) and CT (Thr399Ile) variants were more frequent among patients with chronic CL than patients with the acute form of the disease; however, they observed no significant differences between acute CL patients and asymptomatic cases. 24 In another study, the association between TLR4 gene mutations (A896G and C1196T SNPs) and VL was investigated among the Iranian population by Rasouli et al. 25 In this case, no statistically significant differences were observed in A896G and C1196T alleles and genotypes when VL patients and the controls were compared. Therefore, they concluded that TLR4 gene polymorphisms at the 896 and 1196 positions cannot be regarded as the major contributors to VL susceptibility. Recently, Ejghal et al. also demonstrated the possible association of TLR4 Asp299Gly, TLR4 Thr399Ile and TLR2 Arg753Gln polymorphisms with VL in Moroccan children. 26 They recruited 119 children with VL caused by Leishmania infantum as well as 138 unrelated children, 95 asymptomatic cases and 43 healthy controls who had no indication of present or prior infection. Their results showed significant differences in genotype Thr399Ile and recessive model frequencies between the VL and delayed-type hypersensitivity (DTH+) groups respectively. Concerning the Asp299Gly, there were significant associations when comparing VL versus DTH+ and DTH+ versus DTH- groups. Moreover, there was a significant association regarding the TLR4 (Arg753Gln) genotype, allele frequencies and when applying a recessive model in the VL versus DTH+ groups. They concluded that allele C in Thr399Ile and allele G in Arg753Gln polymorphisms may lead to protection against the clinical disease.
TLR9 SNP interferes with the ligand-binding activity of the receptor and cause defects in signal transduction, leading to impairment in cytokine release from immune cells and alter human immune responses.18,27–30 When we studied the polymorphism in the case of TLR9, we observed that T1237 genotype was more frequent in VL patients (43.3%) than the healthy controls (33.3%) and the difference was statistically significant (P = 0.00391) (Tables 3--5). Our study is consistent with the study by Carvalho et al., where a link between TLR-9 polymorphism (T-1237C) and allergic broncho-pulmonary aspergillosis (ABPA) was demonstrated. 31 The patients with ABPA had a significantly higher frequency of allele C for the T-1237C SNP in TLR-9 than control patients (20.5 versus 9.4%). This polymorphism was located within the putative promoter of the TLR-9 gene and has been implicated in chronic inflammatory diseases, including asthma. Previous reports also suggested TLR9 T1237C SNP disrupts the ligand binding capability of TLR9 and influences the risk of malaria occurrence in children. 28 Another study conducted in Ghana reported that T1237C SNP increased the susceptibility to malaria during the first pregnancy in women. 32 This same SNP was also tested for a possible role in infection risk by human papilloma virus (HPV), but was found not to be associated significantly with either HPV clearance or persistence. 33
Therefore, based on our findings, we suggest the TLR pathway should be extensively explored to understand the molecular mechanism of disease pathogenesis. We would advocate that the knowledge of genetic polymorphisms could be helpful for better treatment options for the management of VL cases. Previous studies reported that, based on the genotype of individual, personalized chemotherapy has been prescribed for cancer patients. Personalized medicine bases the therapy options on a patient’s molecular profile. 34 , 35 Pharmacogenetics is the study of patients’ genotypes affecting drug response. It is often been found that certain drugs work well in some patient groups but not in others. Therefore, studies between the genotype of patients and the response of therapeutics helps in designing more potent and individual-specific chemotherapeutic approach and combat drug-resistance issues. 36
Conclusion
In conclusion, the present study explored the role of TLR4 Asp299Gly, TLR4 Thr399Ile and TLR91237C polymorphisms in Leishmania infection in Bihar state, India. No significant differences in the TLR4 genotype or allele distributions between patient and healthy groups were observed. However, significant difference in TLR9 genotype or allele distributions were observed among VL patients and healthy groups. This study also confirmed the low frequency of TLR4 polymorphisms in our population, as observed in other studies. Although no significant association was found between TLR4 polymorphisms and occurrence of VL, this does not infer a lack of involvement of the same. Studies involving a large population from different independent geographically located or ethnic groups may provide a clear and more comprehensive picture about the involvement of these polymorphisms with Leishmania infection. These may serve as risk factors in estimating the occurrence of VL in the endemic population, thus aiding in its proper management.
Our pilot study aims to understand the probable association between the TLR4/9 polymorphism and VL susceptibility. It is important to mention that due to the active implementation of a kala-azar (VL) elimination programme by the government of India, the cases of VL drastically reduced during the study period. Indeed, recruitment of a higher number of healthy people and patients and more detailed and elaborated studies could provide a better understanding of the relationship of TLR4/TLR9 polymorphisms on the disease manifestation of VL.
Footnotes
Acknowledgements
The authors are grateful to Avdhesh Kumar for his support in some experiments. We are indebted to Naresh Kumar Sinha and Amarkant Singh for providing technical assistance for this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out by the Intramural Fund from ICMR, Ministry of Health and Family Welfare, Government of India. The study was part of an intramural project carried out by Manish Kumar. The project aimed to detect TLR4 and TLR9 polymorphisms in the kala-azar endemic regions of Bihar by Bi-PASA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. AM was supported by a Senior Research Fellowship from the ICMR, Government of India.