
Abstract
Nuclear hormone receptor ligands are known to modulate innate immunity by dampening the immune response induced by pathogens. Here, we report that unlike other ligands, 3,3′,5-triiodo-
Introduction
Host innate immune response triggers the activation of IFN regulatory and NF-κB transcription factors and induces the production of type I IFNs and pro-inflammatory cytokines.1–4 IFNs induce a diverse range of gene products called IFN-stimulated genes (ISGs) that function as effectors of the type I IFN response. 5 The double-stranded RNA (dsRNA)-dependent protein kinase (PKR) is an ISG that is induced and activated during viral infection and plays a key role in type I IFN production via a positive feedback loop response. Activated PKR phosphorylates the α-subunit of translation initiation factor eIF-2, resulting in translational shutdown and inhibition of viral replication. 6 , 7 Thus, PKR plays a central role as an ISG that links IFN response pathways to stress-mediated response during viral infections.
Nuclear hormone receptors play important roles in the development and homeostasis of both adaptive and innate immune responses.8–13 The ligands of these receptors modulate gene expression by directly binding to their cognate receptors, trans-repression involving interaction with other transcription factors, or via non-genomic13–18 mechanisms that may or may not involve the binding of ligands to the receptors and are mostly non-nuclear. A number of nuclear receptor ligands have been reported to have anti-inflammatory activities.
9
These include ligands for glucocorticoid receptor (GR), retinoic acid receptors (RARs), vitamin D receptor (VDRs), peroxisome proliferator-activated receptors (PPARs), estrogen receptor (ER), and liver X receptor (LXR). In contrast, increased levels of thyroid hormone receptor (THR) ligands 3,3′,5-triiodo-
We recently showed that supraphysiological concentrations (SPCs) of T3 induce integrated stress response (ISR) signaling pathways and inhibit viral replication, 23 suggesting a link between ISR and antiviral pathways. In this paper, we studied the effect of T3 on innate immune response pathways and report that the hormone induces IFN response and the expression of ISGs, and this effect is significantly amplified at SPCs and in combination with ds-RNA mimic polyinosinic–polycytidylic acid (Poly IC). In addition, we show that PKR plays a differential role in regulating T3-mediated ISG expression.
Methods
Cells and reagents
HEK293FT (Life Technologies, Grand Island, NY), HEK293 (ATCC, Manassas, VA), HEK-293/TLR3 (provided by Dr. Kamal Saikh, Army Medical Research Institute of Infectious Diseases, Frederick, MD), HeLa (ATCC), MCF-7 (ATCC), RIG-I expressing Huh-7, RIG-I defective Huh-7.5 cells 24 (Apath, Brooklyn, NY), and HeLa/control ShRNA, HeLa/PKR-ShRNA/Kd (provided by Dr. Charles E Samuel, University of California, Santa Barbara, CA) were maintained in DMEM containing 10% FBS. HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells were maintained in the presence of 1.0 µg/ml puromycin. Human PBMCs, obtained by lymphapheresis of healthy donors, were purified by Ficoll density gradient centrifugation. Monocytes were isolated from PBMCs by the adherence technique. 25 Poly IC, all-trans-retinoic acid (ATRA), vitamin D, azelaoyl, T3, cytosporone B, dexamethasone, GW3965, LY294002, U0126, and U73122 were purchased from Sigma–Aldrich (St. Louis, MO). Cyclo (RGDfC) αVβ3 integrin-binding peptide (RGD) and Cyclo (-RADfK-), RGD negative control peptide, were obtained from Anaspec (Fermont, CA). Abs to AKT, phospho-AKT, and PKR were from Cell Signaling Technology (Danvers, MA). Abs to GAPDH and THRα were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX).
Treatment and transfection with Poly IC
HEK293FT, HEK293, and HEK293/TLR3 cells were treated with 50 µg/ml Poly IC on six-well plates in the presence of solvent or nuclear hormone receptor ligands, as indicated. HeLa, HeLa/control ShRNA, HeLa/PKR-ShRNA/Kd, Huh-7, Huh-7.5, and PBMCs were transfected with 5.0 µg Poly IC on six-well plates using Fugene-HD (Promega, Madison, WI) in either the presence or the absence of T3. Cells were harvested after 16 h of treatment for RNA isolation. In other experiments, HEK293FT cells were transfected with 1.0 µg empty vector or cTHRα plasmid for 24 h. Cell were then treated with DMSO or T3 for 16 h in the presence or absence of 50 µg/ml Poly IC and harvested for RNA isolation.
Plasmids and reporter assays
pCAGGS-Flag-RIG-I-N plasmid and empty vector were provided by Dr. Adolfo García-Sastre (Mount Sinai School of Medicine, NY). IFIT1-Promoter-Luc plasmid was kindly provided by Dr. John Hiscott (St. Luce, FL). ISRE-Luc plasmid was obtained from Affymetrix (Santa Clara, CA). TRE-response element containing plasmid TATA-DR4-Luc was constructed by cloning the AGGACAc tcaAGGACAc tcaAGGACActca AGGACA sequence, as described previously. 26 Dr. M Mahajan (NYU Medical Center, NY) provided a plasmid expressing cTHRα. For reporter assays, 50 ng IFIT1-Promoter-Luc or 150 ng of ISRE-Luc plasmids was used for transfection using Fugene-HD. After 24 h, cells were treated with 50 μg/ml Poly IC (HEK293FT) or transfected using Fugene-HD with 5 µg Poly IC (HeLa) in the presence or absence of 75 µM T3. Sixteen h after treatment, cells were harvested for luciferase measurement. In other experiments, HEK293FT cells were transfected with 1.0 µg of TATADR4-Luc in the presence or absence of 1.5 µg empty vector or cTHRα plasmid. Cells were treated with T3 after 24 h of transfection and harvested 16 h later for luciferase activity measurement. Luciferase activity was normalized to protein concentrations in the extracts.
Western blot
Protein extracts were electrophoresed in a 10% or 12% NuPAGE Bis Tris Gel using NuPAGE MES-SDS running buffer (Life Technologies) and transferred to a polyvinylidene fluoride membrane using XCell Blot Module (Life Technologies). After treatment with primary Abs, protein was detected using fluorophore labeled secondary Abs and the Odyssey Infrared Imaging System (Li-cor Biotechnology, Lincoln, NE).
Real-time RT-PCR
Total RNA was isolated using a RNAeasy kit (Qiagen, Germantown, MD) by following the manufacturer’s protocol. For real-time RT-PCR, total RNA was reverse transcribed with Superscript II reverse transcriptase (Life Technologies) in the presence of 2.5 μM of random hexamers. The products of reverse transcription were subject to real-time PCR with gene-specific primers (spanning the introns and designed by NCBI primer-blast tool: https://www.ncbi.nlm.nih.gov/tools/primer-blast/) using the 7500 Fast Real-Time PCR System and Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA). Each reaction included an RT-negative control. The comparative threshold cycle method was used to calculate the relative gene expression. The data were normalized to the geometric mean of three mRNAs—GAPDH, PKR1, and WARS2—the levels of which did not change after the treatments.
Statistical analysis
The results are presented are the mean of three independent experiments (each experiment was performed in two to three replicates), and each individual experimental mean is shown in the figures. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison tests using GraphPad Prism v8 (GraphPad Software, Inc., San Diego, CA). Differences between groups were considered statistically significant if P < 0.05. Statistical significance is denoted with asterisks (*P < 0.05; **P < 0.01; NS = not significant).
Results
T3 hormone induces and amplifies Poly IC-mediated ISG expression
We studied the effect of various nuclear hormone receptor ligands on Poly IC-induced expression of ISGs. Initial studies were carried out with HEK293FT cells, as they were found to be highly responsive to the treatment with Poly IC. We choose to study the expression of IFIT1, IFIT2, IFIT3, and IP10 as examples of Poly IC-induced ISGs. We found that while the treatment of HEK293FT cells with 1 µM of ATRA, vitamin D, azelaoyl, cytosporone B, dexamethasone, or GW3965 (ligands for RAR/RXR, VDR, PPARγ, Nurr77, GR, and LXR, respectively) had either no effect or inhibited Poly IC-induced expression of ISGs to varying degrees (Figure 1a), treatment with 1 µM T3 (ligand for THR) enhanced Poly IC-induced ISG expression. T3 effect on Poly IC-induced ISG expression was dose dependent and surprisingly at SPCs (10–75 µM) T3 significantly amplified Poly IC-mediated ISG expression (Figure 1b), indicating that unlike other nuclear receptor ligands that dampen the ISG expression, T3 has an amplifying effect on innate immune response induced by dsRNA viral mimic Poly IC. T3 alone showed moderate induction of ISGs that was also dose-dependent and was highest at 75 µM (Figure 1c). To study if T3 also induced ISG expression in physiologically relevant non-transformed cells, monocytes isolated from PBMCs were transfected with Poly IC followed by treatment with DMSO or 75 µM T3. RT-PCR analysis for IFIT1, IFIT2, and IFIT3 mRNAs revealed that T3 significantly amplified Poly IC-mediated expression of these ISGs (Figure 1d) in addition to inducing moderate expression in the absence of Poly IC (Figure 1e). In addition to PBMCs, we also found that SPCs of T3 amplified Poly IC-mediated expression of ISGs in a number of other cell types, including MCF-7 (data not shown), HeLa, HEK293, and Huh-7 (see below), indicating that T3 induces IFN response and the induction of ISGs broadly across different cell types.
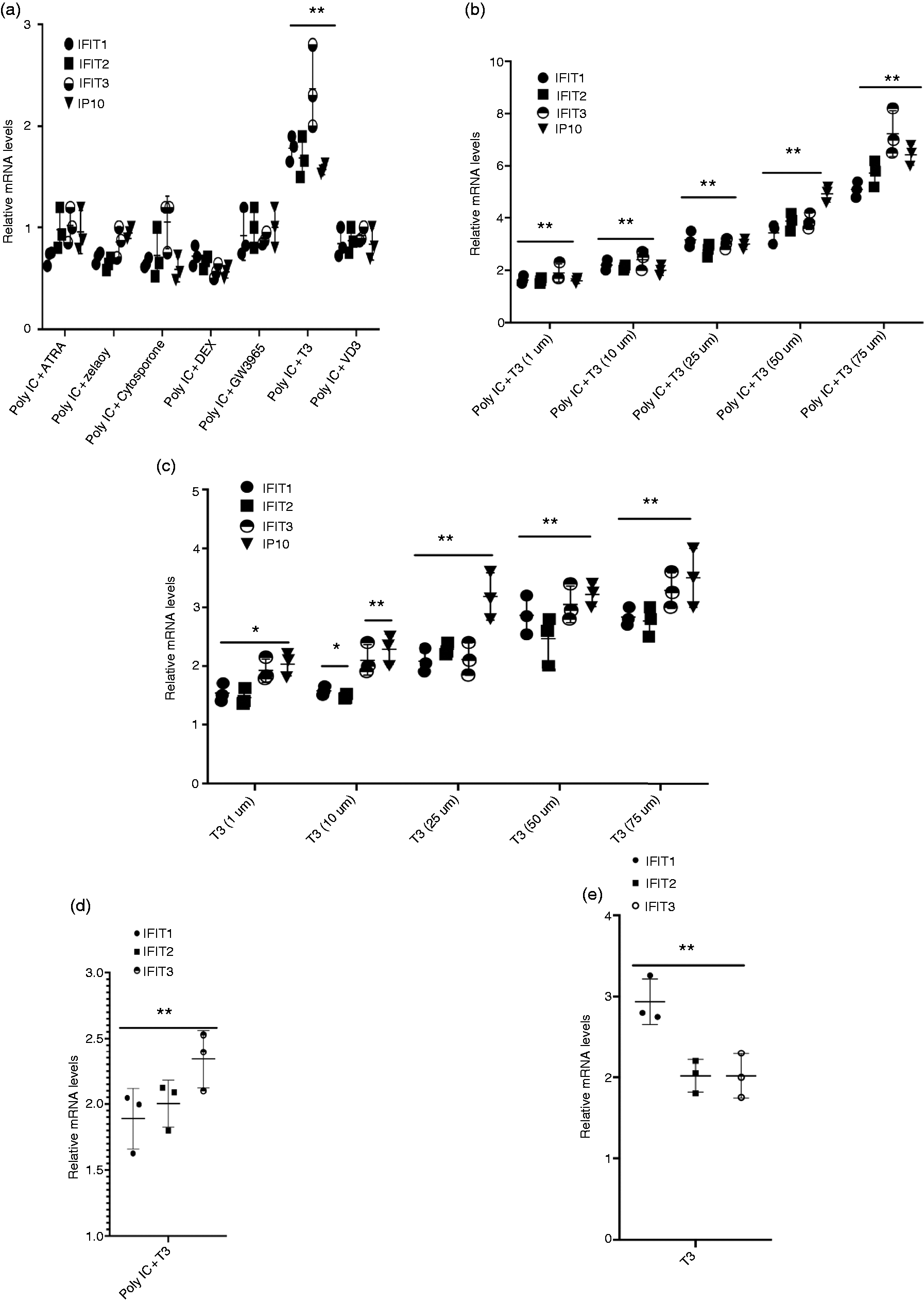
Thyroid hormone 3,3′,5-triiodo-
T3 induces ISG expression by TLR3-, RIG-I-, and IFN-β1-independent mechanisms
TLR3 and RIG-I are two highly studied dsRNA-binding sensors essential in innate immune response mediated by RNA viruses and dsRNA mimic Poly IC. 2 , 3 , 27 , 28 To study the role of TLR3 and RIG-I in T3-mediated Poly IC-induced ISG expression, we used TLR3-overexpressing HEK293, TLR3-low expressing HEK293, RIG-I expressing Huh-7, and RIG-I defective Huh-7.5 cells. 24 Cells were treated (HEK-293) or transfected (Huh-7 and Huh-7.5 cells) with Poly IC in the presence or absence of DMSO or 75 µM T3. RT-PCR for IFIT3 mRNA, a representative of an ISG that is significantly induced by T3 in combination with Poly IC (Figure 1b), revealed that the effect of T3 on Poly IC-mediated expression was not affected by the presence or absence of TLR3 (Figure 2a) or RIG-I (Figure 2b). These results indicate that the expression of TLR3 or RIG-I is not critical for T3-mediated induction of IFIT3 by Poly IC. In addition, the induction of IFIT3 by T3 alone was also independent of TLR3 or RIG-I expression (Figure 2a and b). RIG-I-independent expression of ISGs by T3 was further confirmed by studying the effect of T3 on RIG-I-induced ISG expression. Expression of a carboxy-terminal truncated RIG-I (RIG-I-N) containing the CARD and lacking the helicase domain of RIG-I is known to activate the IFN-β response and induce ISG expression in the absence of Poly IC. 29 293FT cells were transfected with either vector or RIG-I-N plasmid in the presence of DMSO or T3. As expected, the transfection of RIG-IN plasmid resulted in a marked expression of IFIT3 (Figure 2c). However, T3 did not further induce the expression of IFIT3 in these cells (Figure 2d). Surprisingly, T3 at 75 µM inhibited the expression of IFIT3 by nearly 50%. These results provide further evidence that T3-mediated expression of ISG is RIG-I independent.
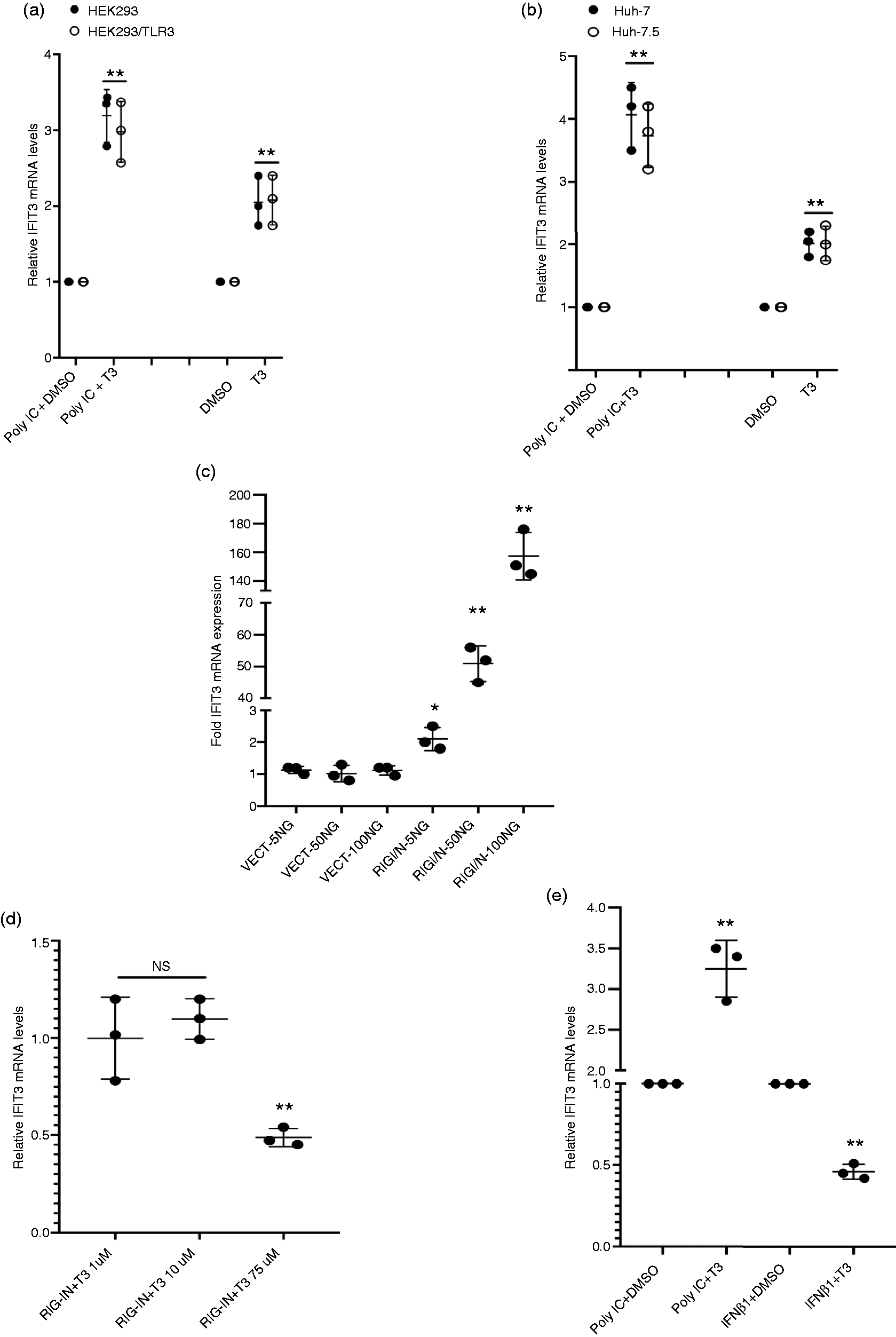
T3 induces ISG expression by TLR3-, RIG-I-, and IFN-β1-independent mechanisms. (a) TLR3-low expressing HEK293 and TLR3-overexpressing HEK293/TLR3 cells were treated with 50 ug/ml Poly IC in the presence or absence of DMSO or 75 µM T3 for 16 h. (b) RIG-I expressing Huh-7 and RIG-I defective Huh-7.5 cells were transfected with Poly IC in the presence or absence of DMSO or 75 µM T3 for 16 h. (c) 293FT cells were transfected with indicated concentrations of empty vector or RIG-IN plasmids for 36 h. (d) 293FT cells were transfected with 100 ng of indicated plasmids for 24 h and then treated with DMSO or indicated concentrations of T3 for 16 h. (e) 293FT cells were treated for 16 h with DMSO or 75 µM T3 in the presence or absence of Poly IC or 1000 μg/ml IFN-β1. RNA was isolated from the cell pellets and the expression of IFIT3 quantitated by real-time RT-PCR as described in the Methods section. Wherever applicable, the data presented are in comparison to DMSO control groups. *P < 0.05; **P < 0.01; NS: not significant.
We next explored the effect of T3 on IFN-β1-induced IFIT3 expression. RT-PCR analysis showed that the treatment of HEK293FT cells with IFN-β1 induced a robust expression of IFIT3 (Figure 2e) and other ISGs (data not shown). Although treatment with T3 alone induced moderate expression of IFIT3, it did not amplify IFN-β1-induced ISG expression. Instead, there was a moderate inhibitory effect of T3 on IFN-β1-induced ISG expression, suggesting that T3 does not induce ISG expression using IFN-β1-dependent pathways. Together, these results indicate that T3 significantly amplifies Poly IC-mediated induction of ISGs using pathways that are independent of the presence of TLR3 and RIG-I, and IFN-β1.
T3 hormone induces ISG expression using non-genomic mechanisms involving integrin αVβ3 binding and activation of PLC–phosphatidyl-inositol 3-kinase–AKT pathways
T3 modulates gene expression either by genomic mechanisms involving direct binding to THR heterodimerized with RXR on the promoters of target genes or via non-genomic mechanisms that may or may not involve the binding of T3 to THR and is mostly non-nuclear.13–18, 30 , 31 To understand the mechanism of T3-induced ISG expression, we analyzed the promoters of the ISGs studied here for the presence of classical THR binding sequences (TRE) and found that none of them contained any known TREs, indicating that genomic mechanisms may not be involved in the ISG induction. We found that HEK293FT cells expressed low levels of THR (Figure 3a) that was not sufficient to activate TKDR4-Luc reporter plasmid following T3 treatment (Figure 3b). However, if these cells were transfected with THRα plasmid, a robust activation of TKDR4-Luc was observed after treatment with T3 (Figure 3b). To study if the overexpression of 293FT cells with THRα will also influence the levels of ISG induction by Poly IC following T3 treatment, cells were transfected with THRα expressing plasmid for 24 h followed by treatment with Poly IC in the presence and absence of T3. The data show that THRα expression has no effect on the T3-mediated induction of IFIT3 (Figure 3c). Further evidence that genomic mechanism involving the binding of T3 to THR on ISG promoter was not involved in T3 action was obtained by performing transient transfection assays using plasmids containing IFN-stimulated response elements (ISRE). ISRE-Luc or IFIT1 promoter-Luc reporter plasmids with ISRE are known to be induced in response to Poly IC treatment. HEK293 cells were transfected with the reporter plasmids in the presence of vector or THRα and then treated with T3 either alone or in combination with Poly IC. As expected, treatment with Poly IC induced the luciferase activity from the reporter vectors (Figure 3d). However, T3 did not further induce luciferase activity either alone or in combination with Poly IC and in either the presence or the absence of THRα. Similar results were obtained with HeLa cells that do not express functional THR 32 (data not shown). If treatment with T3 would involve binding of T3 to THR on the ISG promoters, then induction in the luciferase activity was expected following treatment with T3. Together, these findings indicate that genomic mechanisms involving binding of T3 to THR on ISG promoters may not be involved in the T3-induced ISG induction.
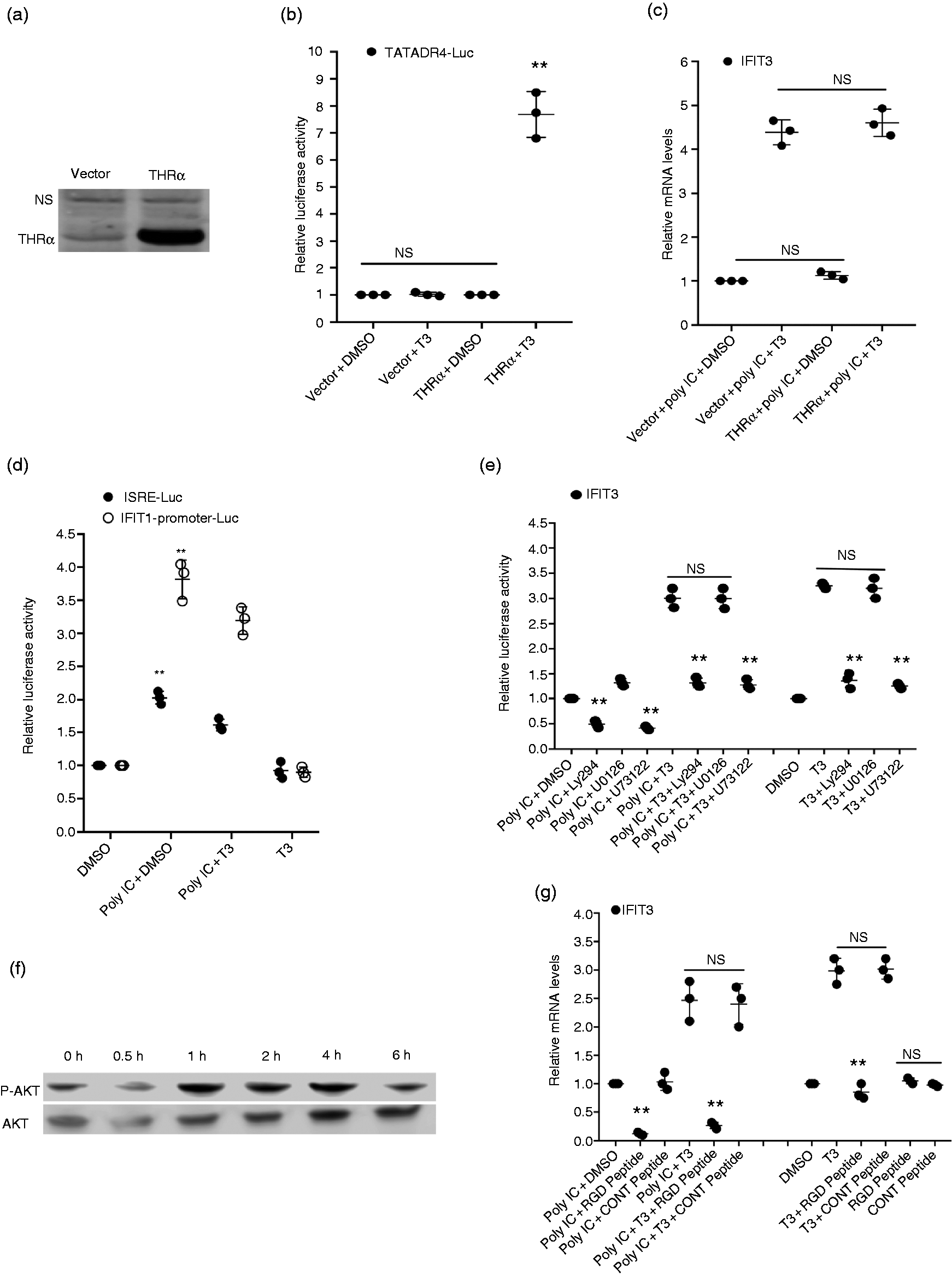
T3 induces ISG expression using non-genomic mechanisms involving integrin αVβ3 binding and activation of PLC–PI3K–AKT pathways. (a) HEK293FT cells were transfected with 1.5 µg of empty vector or cTHRα plasmid and protein extracts were processed for Western blot using Abs to THRα/β. Non-specific (NS) band is shown for equal loading. (b). HEK293FT cells were transfected with 1.0 μg of TATADR4-Luc in the presence or absence of 1.5 µg of empty vector or cTHRα plasmid. After 24 h of transfection, cells were treated with 75 µM T3 and harvested 16 h later for luciferase activity measurement. **P < 0.01. NS: not significant. (c) HEK293FT cell were transfected with 1.0 µg of empty vector or cTHRα plasmid for 24 h. Cells were then treated with DMSO or 75 µM T3 for 16 h in the presence or absence of Poly IC. (d) 50 ng of IFIT-Promoter-Luc or 100 ng ISRE-Luc plasmids were transfected in HEK293FT cells followed by treatment with Poly IC in the presence or absence of DMSO or 75 µM T3. Cells were harvested and luciferase activity was measured. **P < 0.01. (e) HEK293FT cells were treated with T3 (75 µM) or Poly IC either alone or in combination with each other in the presence of either DMSO or LY294002 (5 µM), U73122 (10 µM), or U0126 (5 µM) for 16 h. (f) HEK293FT cells were treated with T3 (75 µM) for indicated time periods. Proteins extracts were analyzed by Western blot using Abs to total AKT and phospho-AKT. (g) HEK293FT cells were treated for 16 h with DMSO or 75 µM T3 in the presence or absence of Poly IC and 50 µg/ml RGD peptide or a control peptide. RNA was isolated from the cell pellets (c, e, and g) and the expression of IFIT3 quantitated by real-time RT-PCR as described in the Methods section. All the data presented are in comparison to DMSO control groups. **P < 0.01. NS: not significant.
Numerous studies have shown that T3 induces non-genomic effects on gene expression by binding to integrin αVβ3 and activating phosphatidyl-inositol 3-kinase (PI3K)–AKT pathways.14–18 To test if T3 induction of ISGs was dependent on the activation of PI3K, we treated HEK293FT cells with Poly IC and T3 in the presence of LY294002, an inhibitor of PI3K. The results showed that LY294002 inhibited IFIT3 expression induced by T3, Poly IC, and a combination of T3 and Poly IC (Figure 3e). A phospholipase C (PLC) inhibitor U73122 similarly inhibited the induction of IFIT3 by Poly IC and T3. In contrast, MAPK inhibitor U0126 did not inhibit the expression of IFIT3 induced by Poly IC, T3, or Poly IC+T3. One of the downstream targets of the PI3K activation is the phosphorylation of AKT. To examine whether T3 activated the AKT pathway, we tested the effect of T3 on the phosphorylation of AKT. Cells were treated with 75 µM T3 for different time periods ranging from 30 min to 6 h, and the activation of AKT was monitored by Western blot with Abs to total and phospho-AKT. Results showed that AKT phosphorylation was significantly induced within 1 h (Figure 3f) and remained high until 4 h treatment and decreased to near basal level by 6 h. Overall, these data suggest that T3 induces the activation of PI3K that in turn activates downstream AKT pathways, implicating PI3K-dependent AKT activation in the amplification of IFN response by T3.
To explore the role of integrin binding by T3 for its ability to induce ISG expression, we used RGD (Arg-Gly-Asp) peptide to block the integrin binding sites. RGD peptide is known to mimic the cell adhesion proteins and bind integrin αVβ3. 33 HEK293FT cells were treated with T3 in the presence or absence of Poly IC and RGD or a control peptide. Unlike control peptide, treatment with RGD peptide, which blocks the binding of T3 to integrin αVβ3, was found to abrogate the induction of IFIT3 by T3 and Poly IC either alone or in combination with each other (Figure 3g). Similar results were obtained with RT-PCR using primers for other ISGs and IFN-β1 (data not shown). These data indicate that binding of T3 to αVβ3 integrin is crucial for both T3- and Poly IC+T3-induced ISG expression. Overall, these data suggest that T3 induces the ISG expression using non-genomic mechanisms involving integrin αVβ3 binding and the activation of PLC–PI3K–AKT pathways.
PKR selectively regulates the T3-induced expression of ISGs
PKR is an ISG that is induced and activated during viral infection and plays a key role in type I IFN production. To study the role of PKR in T3-induced ISG expression, we used HeLa/PKR-ShRNA/Kd cells in which PKR protein (Figure 4a) and mRNA (Figure 4b) expression is knocked down by > 90% by stable expression of PKR-specific ShRNA. 34 HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells were treated with T3 (75 µM) for 16 h, and the expression of a panel of 81 known ISGs were studied by RT-PCR. Table 1 shows the list of 24 genes that were induced by 1.5-fold or more in HeLa/control ShRNA or HeLa/PKR-ShRNA/Kd following treatment with T3. Further analysis revealed that the presence of PKR differentially modulated the expression of these T3-induced ISGs. Strikingly, PKR knockdown increased the T3-induced expression of 15/24 ISGs and inhibited the expression of 5/24 T3-induced ISGs. These were classified as PKR-inhibited and PKR-dependent ISGs, respectively (Table 1). Notable among the PKR-inhibited ISGs were the cytokines CCL17, CXCL11, and IL-6 in addition to ISGs such as IFIT1, IFIT2, IFIT3, RSAD2, TNF, and IRF1. Among the PKR-dependent T3-induced ISGs were GEM, IFN-β1, and MMP13 whose induction was highly inhibited in PKR knockdown cells. Induction of 4/24 T3-mediated ISGs either remained unchanged or was only moderately affected by PKR knockdown (PKR independent). These included TLR3, MEFV, IL-1A, and IFIT5 (Table 1).
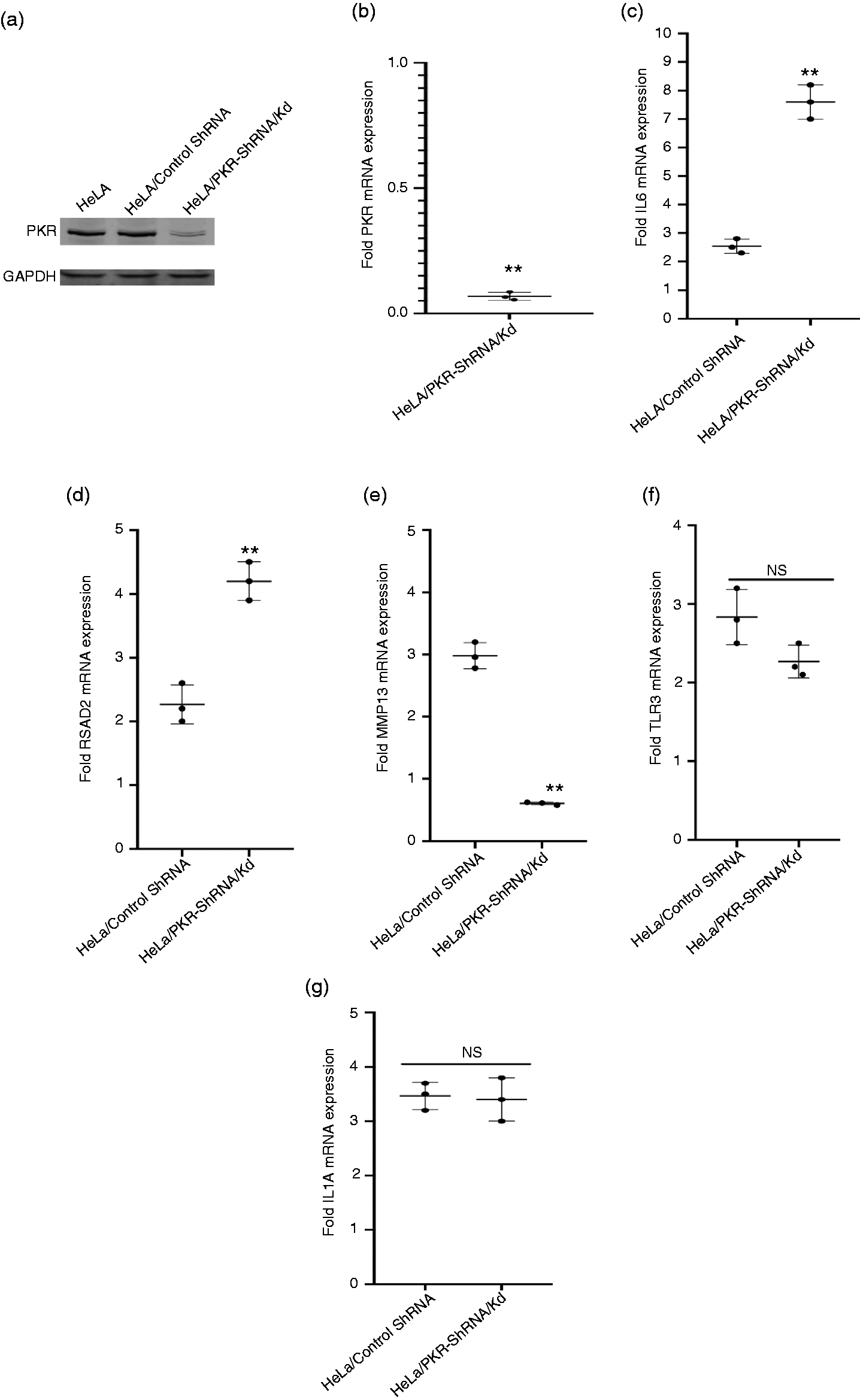
RNA-activated protein kinase (PKR) selectively regulates the T3-induced expression of ISGs. (a) Protein extracts were prepared from HeLa, HeLa/control ShRNA, and HeLa/PKR-ShRNA/Kd cells and probed with Abs to PKR and GAPDH. (b) Total RNA was isolated from HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells, and the expression of PKR mRNA was quantitated by RT-PCR as described in the Methods section. The data presented are in comparison to PKR mRNA levels in HeLa/control ShRNA cells. (c)–(g) HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells were transfected with Poly IC in the presence of DMSO or 75 µM T3 for 16 h. RNA was isolated from the cell pellets, and the expression of indicated genes was quantitated by RT-PCR as described in the Methods section. The data presented are in comparison to T3+DMSO control groups. **P < 0.01. NS: not significant.
List of genes induced by T3 in HeLa/control ShRNA or HeLa/PKR-ShRNA/Kd cells.
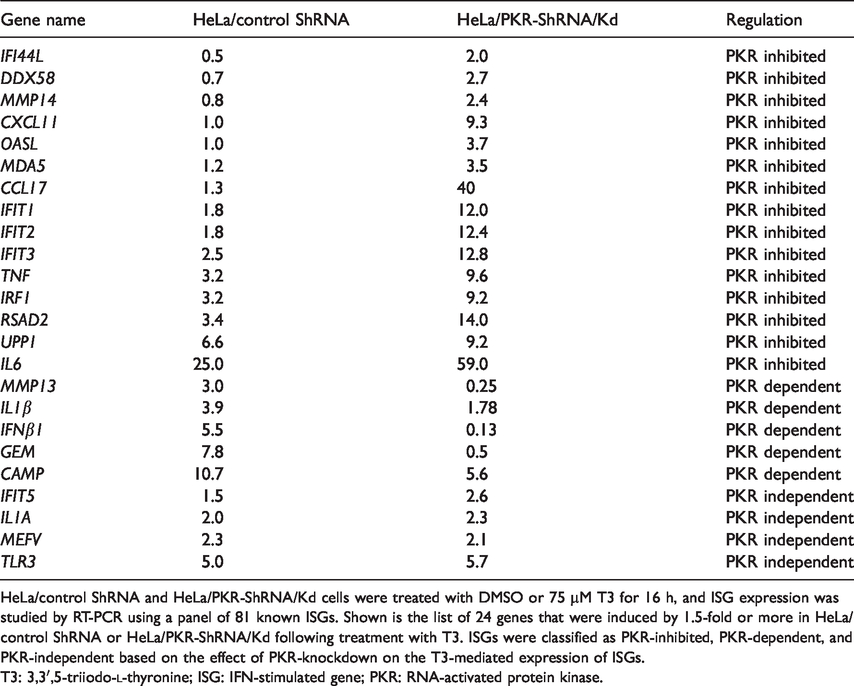
HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells were treated with DMSO or 75 µM T3 for 16 h, and ISG expression was studied by RT-PCR using a panel of 81 known ISGs. Shown is the list of 24 genes that were induced by 1.5-fold or more in HeLa/control ShRNA or HeLa/PKR-ShRNA/Kd following treatment with T3. ISGs were classified as PKR-inhibited, PKR-dependent, and PKR-independent based on the effect of PKR-knockdown on the T3-mediated expression of ISGs.
T3: 3,3′,5-triiodo-
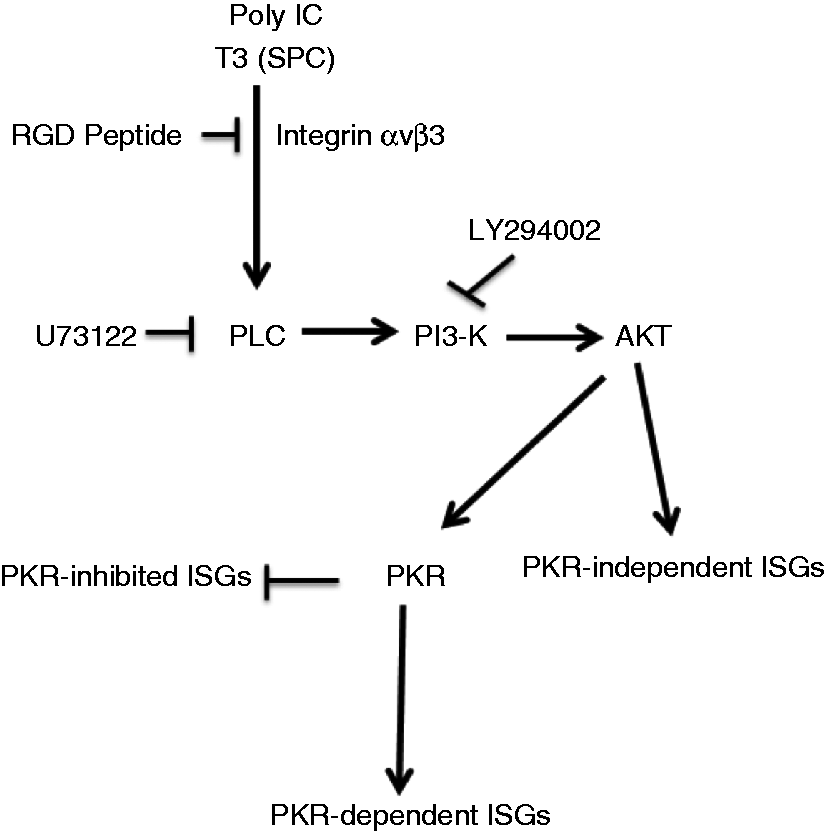
Proposed signaling pathways for T3-mediated induction of innate immune response. T3 at SPCs activates innate immune response by non-genomic mechanisms, involving integrin binding and PLC–PI3K–AKT pathways. PKR plays an essential role in T3 function by selectively modulating T3-induced ISG expression.
To study the role of PKR in T3-mediated amplification of Poly IC-induced ISG expression, HeLa/control ShRNA or HeLa/PKR-ShRNA/Kd cells were transfected with Poly IC in the presence of DMSO or 75 µM T3. As with T3 alone, the presence of PKR also differentially modulated the ISG expression induced by a combination of Poly IC and T3. Whereas IL-6 (Figure 4c) and RSAD2 (Figure 4d) were amplified more efficiently in the PKR knocked-down cells compared to control cells, T3 failed to amplify the Poly IC-induced expression of MMP13 (Figure 4e) in PKR knocked-down cells. The amplification of TLR3 (Figure 4f) and IL-1A (Figure 4g) expression was not affected by the PKR knockdown. Together, these data reveal that PKR plays a selective role not only in regulating the T3-induced expression of ISGs but also in the amplification of ISG expression mediated by a combination of Poly IC and T3.
Discussion
Our data provide evidence that the thyroid hormone T3 induces IFN response and the expression of ISGs, and this effect is significantly amplified at supra-physiological concentrations and in combination with the dsRNA mimic Poly IC. This finding underscores the unique functional aspects of T3 as an inducer of an innate immune response. Innate immune responses are generally not restricted to a specific cell type and can be elicited in a variety of cell types. 35 Because a number of cell lines and human primary monocytes were found to respond to T3, the data suggest that T3 at SPCs could induce an IFN response and the induction of ISG broadly across different cell types. Poly IC-mediated ISG expression and T3-mediated amplification of Poly IC-induced ISG response were independent of the presence of TLR3 and RIG-I, two sensors that are known to bind dsRNA, indicating that TLR3 and RIG-I are not essential for Poly IC and T3 action. However, this does not exclude the possibilities of involvement of other known or unknown cytoplasmic sensors in the T3 action. In addition, T3-induced ISG response amplification was not seen upon IFN-β1 treatment, ruling out the role of the IFN-β–IFNR signaling pathway in the T3 action.
A lack of TRE sites in the promoters of ISGs and the unresponsiveness of THR overexpression to T3-induced ISG expression indicated that the presence or the absence of THR is not important for T3-mediated ISG induction and pointed to the possibility of a non-genomic mechanism operating in the regulation of T3-mediated ISG induction. The non-genomic effects of T3 have been defined in a number of previous reports.14–18 These mostly include the outcomes that are seen at physiological concentrations of T3. Here, we found that SPCs of T3 are needed to induce ISGs, indicating that another level of non-genomic effect operates at the SPCs of T3 that results in ISG induction. However, our studies do not rule out the importance of some unknown indirect genomic mechanisms that may be regulating T3 function. Using inhibitors of ERK, PI3K, and PLC activation and a peptide that interferes in integrin binding, we have shown the importance of integrin binding, calcium mobilization, and PI3K pathways in the induction of ISGs by T3. In addition, treatment with T3 induced AKT phosphorylation, indicating a PI3K–AKT nexus in regulating T3 function.
ShRNA knockdown data provided evidence that PKR, an ISG that is induced and activated during viral infections, plays an important regulatory role in T3-induced expression of ISGs and the amplification of ISG expression mediated by a combination of Poly IC and T3. PKR-modulated ISGs were classified as PKR-inhibited, PKR-dependent, and PKR-independent.
The mechanism involved in PKR-mediated regulation of T3 function is not yet clear. We have recently shown that the SPCs of T3 amplify Poly IC-mediated expression and activation (phosphorylation) of PKR 23 and thus may have an indirect role in regulating PKR-mediated ISG expression. In the same study, we also reported that SPCs of T3 induce ISR signaling pathways and inhibit vesicular stomatitis viral replication, suggesting a link between ISR and antiviral pathways. We have also reported that the overexpression of an ISR-mediated protein GADD34, also an ISG, 23 inhibits HIV-1 replication, 36 further confirming the crosstalk between ISR and innate immune response pathways. Future studies should focus on the mechanisms involved in the T3-mediated crosstalk between these two pathways and study the importance of such interactions in modulating pro-inflammatory and antiviral responses. A recent report using mice showed that physiological concentrations of T3 can clear meningococcal infections. 37 Sustained release treatment with T3, where patients were treated orally with T3 every 12 h, has been used in the treatment of symptoms resembling chronic fatigue syndrome. 38 Patients with thyroid disorders are more susceptible to depression, and thyroid hormone supplements have shown enhancement in the clinical response to anti-depressant drugs in depression and prophylaxis-resistant psychiatric illnesses.39–41 Supraphysiological doses of T3 have also been shown to result in a dose-dependent mass loss and reduction in the adiposity in animals in addition to genotoxicity and oxidative stress. 42 , 43 Our current study ascribes another role to T3 and shows that this hormone can also modulate innate immune responses at SPCs.
To summarize, we have uncovered a novel function of T3 in this study and show that T3 at SPCs activates an innate immune response by non-genomic mechanisms, involving integrin binding, calcium mobilization, and PI3K–AKT pathways. We also found that PKR plays an essential role in T3 function by selectively modulating T3-induced ISG expression (Figure 5). These findings have important implications in the basic understanding of the mechanisms of T3 function at SPCs and crosstalk involved in thyroid hormone function and innate immune responses.
Footnotes
Acknowledgements
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This work was supported by the National Institute of Allergy and Infectious Diseases. We are grateful to Drs. Kamal U. Saikh (Army Medical Research Institute of Infectious Diseases, Frederick, MD) for providing HEK-293/TLR3 cell line, John Hiscott (Vaccine & Gene Therapy Institute of Florida, St. Lucie, FL) for IFIT1 promoter-Luc plasmid, Charles E. Samuel (University of California, Santa Barbara, CA) for HeLa/control ShRNA and HeLa/PKR-ShRNA/Kd cells, and Adolfo García-Sastre (Mount Sinai School of Medicine, NY) for providing RIG-IN plasmid.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E.