
Abstract
Abstract
Idiopathic pulmonary fibrosis is a progressive interstitial pneumonia characterised by fibroblast accumulation, collagen deposition and extracellular matrix (ECM) remodelling. It was reported that Akt1 mediated idiopathic pulmonary fibrosis progression through regulating the apoptosis of alveolar macrophage, while its effect on macrophage-produced cytokines remains largely unknown. In the present study, we first examined the phosphorylation of Akt1 in lung sections from idiopathic pulmonary fibrosis patients by immunohistochemistry before applying a bleomycin-induced idiopathic pulmonary fibrosis model using Akt1−/− mice and Akt1+/+ littermates. The results showed that Akt1 was remarkably up-regulated in idiopathic pulmonary fibrosis patients, while in vivo studies revealed that Akt1-deficient mice had well-preserved alveolar structure and fewer collagens, secreted fewer matrix components, including alpha smooth-muscle actin and fibronectin and survived significantly longer than Akt1+/+ littermates. Additionally, the pro-fibrogenic cytokine IL-13 was down-regulated at least twofold in Akt1−/−mice compared to the Akt1+/+group on d 3 and 7 after bleomycin treatment. Furthermore, it was found that Akt1–/– macrophages displayed down-regulation of IL-13 compared to Akt1+/+ macrophages in which Akt1 was phosphorylated in response to IL-33 stimulation. These findings indicate that Akt1 modulates pulmonary fibrosis through inducing IL-13 production by macrophages, suggesting that targeting Akt1 may simultaneously block the fibrogenic processes of idiopathic pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease with worsening of lung function, gas exchange and increased mortality rate approaching 50% of patients.1,2 With this complicated, multistage and progressive process of IPF, its full aetiology is still incompletely understood with limited therapeutic strategies.
During pulmonary fibrosis, various immune cells infiltrate the lung tissues, which release a wide range of inflammatory mediators that regulate inflammation and fibrosis. 3 The role of the Th2 cytokine, IL-13, in pulmonary fibrosis has been widely demonstrated by both in vivo and in vitro experiments. IL-13 transgenic mice were confirmed to lead directly to pulmonary fibrosis pathology, with blocking or germ-line deletion of IL-13 reducing collagen deposition following exposure to bleomycin (BLM).4–6 Clinically, inhibition of human IL-13 with the selective human IgG4 mAb tralokinumab ameliorated established pulmonary fibrosis in a humanised severe combined immunodeficiency disease (SCID) mouse model of IPF. 7
A mechanism study showed that the biological role of IL-13 is partially due to the pro-fibrotic effects of IL-4, as well as through its own receptor (IL-13Rα2).8–10 Together, these studies highlight the role of IL-13 in pulmonary fibrosis. Macrophages have been implicated as central cells in the pathogenesis of IPF. 11 They are recruited by chemokines derived from lung epithelium and participate in the pathogenesis of the disease by secreting cytokines that induce inflammation.12,13 They also serve to produce pro-fibrotic cytokines to induce the proliferation and activation of fibroblasts and promote epithelial-to-mesenchymal transition.14–17 Recently, it was shown that IL-13 produced by macrophages plays a pivotal role in the homeostatic control of the normal lung and the pathogenesis of pulmonary fibrosis.18–20 Clinically, IL-13 expression has been reported in human alveolar macrophages and in bronchoalveolar lavage fluid (BALF) from IPF patients.8,9 In addition, it has been reported that the human neutrophil elastase induces the production of IL-13 in pulmonary fibrosis. However, until now, the regulatory mechanism of IL-13 in macrophages has not been fully estimated.
Akt, also known as protein kinase B, has three isoforms: Akt1, Akt2 and Akt3. As a pro-survival kinase, Akt is activated by numerous stimuli and is implicated in a variety of cellular functions, such as survival, metabolism, transcription and translation. It has also been shown that the PI3K/Akt signalling pathway plays a regulatory role in cancers and inflammatory diseases. In mammalian cells, both Akt1 and Akt2 are widely distributed in the lung tissue, including epithelial cells, fibroblasts and alveolar macrophages. However, its role in pulmonary disease has not been well studied. Previously, our study showed that Akt2 mediates the pathogenesis of pulmonary fibrosis through regulating IL-13 and TGF-β production of macrophages. 21 It was also reported that conditional deletion of Akt1 in mouse alveolar macrophages significantly alleviated BLM-induced IPF through mediating apoptosis resistance. 22 However, the role of Akt1 in modulating the producing pro-fibrotic mediators remains to be elucidated. The present study was conducted to investigate the effect and underlying mechanism of Akt1 on IPF progression.
Materials and methods
Mice
Akt1-deficient mice (Akt1−/−) were kindly supplied by Dr Nissim Hay (University of Illinois at Chicago) and were backcrossed for more than 11 generations on the C57BL/6 strain background. Sex- and age-matched littermates were used in the experiments and were housed under specific pathogen-free conditions at the laboratory animal centre of Shanghai Jiao Tong University (Shanghai, PR China). The procedures were carried out in accordance with the Institutional Animal Care and Use Committee at Shanghai Jiao Tong University.
Preparation of bone marrow–derived macrophages
Femoral and tibia bone marrow cells were isolated from Akt1+/+ and Akt1−/− mice, as previously described.23, 24 ACK buffer (10 mM KHCO3, 150 mM NH4Cl, 0.1 mM EDTA, pH 7.4) was added to the mouse bone-marrow cells flushed from the femurs and tibias to remove erythrocytes before being washed with Ca2+/Mg2+-free PBS and cultured in DMEM supplemented with 10% L929 cell-conditioned medium, 10% FBS and 1% penicillin/streptomycin for 7–9 d. Then, bone marrow–derived macrophages (BMDMs) were stimulated with recombinant murine mIL-33 (10 ng/ml; PeproTech, Rocky Hill, NJ), and PBS was used as control.
BLM-Induced pulmonary fibrosis mice model
Six- to eight-wk-old male Akt1+/+ and Akt1−/− mice (n = 6–8/group) were anaesthetised with an intraperitoneal injection of 5 mg/ml pentobarbital and intratracheally injected with BLM sulfate (BIOTANG, Inc., Lexington, MA) at 1.5 IU/kg for survival and 1.4 IU/kg for other analysis. Intratracheally injected saline was used as control. Lungs were collected for analysis on d 3, 7 and 21. The protein level was determined by Western blot on d 21 and mRNA expression by real-time RT-PCR on d 3, 7 and 21. For histology samples, lungs were perfused with saline and inflated with 4% paraformaldehyde. Sections were stained with hematoxylin and eosin (H&E), Masson’s trichrome and immunohistochemistry.25,26 For RT-PCR and Western blot, sections were preserved in liquid nitrogen.
Lung histology and analysis
On d 3, 7 and 21 after BLM administration, the left lungs (n = 6–8/group) were excised and fixed in 4% paraformaldehyde under a constant pressure. Tissues were then embedded in paraffin blocks, cut into sections 4 µm thick and stained with H&E or Masson’s trichrome (Nanjing Jiancheng Bioengineering Institute, Nanjing, PR China) according to the manufacturer’s instructions. In order to analyse fibrotic changes, a quantitative fibrotic scale (Ashcroft scale) was used. 27 A numerical fibrotic score (Ashcroft scale) was obtained as follows: the severity of the fibrotic changes in each lung section was given as the mean score from the observed microscopic fields. More than 25 fields within each lung section were observed at a magnification of 200×, and each field was assessed individually for severity and allotted a score from 0 (normal) to 8 (total fibrosis). The severity scores for each field were averaged and are presented as the average for each lung section. To avoid bias, all histologic specimens were evaluated in a blinded fashion. Each specimen was scored independently by two observers, and the mean of their individual scores was taken as the fibrotic score.
BALF
BALF was collected by lavaging the lung twice with 1 ml PBS. After the combined samples were centrifuged at 300 g for 5 min, the supernatants were used for total protein analysis using the BCA protein assay kit (Beyotime, Shanghai, PR China), and the pellets were used for the determination of total cell numbers using a blood counting chamber.
Myeloperoxidase (MPO) was measured as an indicator of neutrophil infiltration in response to pulmonary inflammatory injury using tetramethylbenzidine, as previously described. 28
Isolation of total RNA and real-time PCR
RNA was purified from tissue samples or BMDMs using the Tissue Lyser system (Qiagen, Valencia, CA), according to the manufacturer’s instructions. cDNA was prepared using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA) and amplified by real-time RT-PCR with Fast SYBR Green master mix on a Prism 7900HT (Applied Biosystems) with the following primer sets: alpha smooth-muscle actin (α-SMA), forward 5′-GACGCTGAAGTATCCGATAGAACACG-3′, reverse 5′-CACCATCTCCAGAGTCCAGCACAAT-3′; fibronectin, forward 5′-TCTGGGAAATGGAAAAGGGGAATGG-3′, reverse 5′-CACTGAAGCAGGTTTCCTCGGTTGT-3′; IL-13, forward 5′-GAATCCAGGGCTACACAGAAC-3′, reverse 5′-AACATCACACAAGACCAGACTC-3′; and GAPDH, forward 5′-
Western blot
Lung sections from Akt1+/+ and Akt1−/− mice treated with saline or BLM for 21 d were lysed in RIPA buffer containing protease inhibitors (Sigma–Aldrich, St Louis, MO). BMDMs were plated on six-well plates at 2 × 106 cells/well overnight and challenged with 10 ng/ml IL-33 or PBS for 30 min, 1 h and 2 h. Cells were then collected and were lysed in RIPA buffer containing protease inhibitors (Sigma–Aldrich). Protein concentrations were estimated by using the BCA protein assay (Thermo Fisher Scientific, Waltham, MA). Protein lysate was run through 10% Bis-Tris protein gels. The lung tissue lysate was analysed using Abs against α-SMA and β-actin, and the cell lysate was analysed using Abs against Akt, phospho-Akt (Ser473), phospho-Akt1 (Ser473), phospho-p65, p65 and β-actin (Cell Signaling Technology, Danvers, MA). The intensity of Western blot bands was quantified by means of densitometry with ImageJ software (National Institutes of Health, Bethesda, MD).
Immunohistochemistry
Lung sections of IPF patients were kindly provided by Wuxi People’s Hospital, Jiangsu, PR China and were approved by the Institutional Review Board at Wuxi People’s Hospital. Lung sections from murine IPF models and patients (4 µm) were deparaffinised. Endogenous peroxidase was blocked with 1% H2O2 in for 10 min, and nonspecific binding was blocked with 5% normal BSA for 30 min. The slices were incubated with phospho-Akt1 (Ser473; 1:200, Cell Signaling Technology) at 4°C overnight, developed with streptavidin biotin complex kits and visualised with diaminobenizidine. The sections were observed and analysed using a BX60 microscope (Olympus, Tokyo, Japan).
Statistical analysis
Data are shown as the mean ± SEM from at least three independent experiments. Differences between groups of mice were subjected to one-way ANOVA between indicated groups. Student’s t-test was used for other analyses. Survival analysis was performed using log-rank (Mantel–Cox) test. P-values of > 0.05 were considered statistically significant.
Results
Akt1 is phosphorylated in lung tissue from IPF patients
It has been reported that conditional deletion of Akt1 in mouse alveolar macrophages significantly alleviated BLM-induced IPF through mediating apoptosis resistance. 22 In the present study, we first explored the activation of Akt1 in lung tissues from pulmonary fibrosis patients. As shown in Figure 1a, there was a marked increase in the phosphorylation level of Akt1 (Ser473) in fibrosis patients compared to that in normal donors. We also observed increased activation of Akt1 in lung tissue from BLM-challenged mice on d 21 (Figure 1b). Interestingly, all Akt1 phosphorylation positive cells were macrophage-like in shape, which is similar to the phosphorylation level of Akt2. 21 One of the limitations of the methodology is the reliance on only morphological detection of macrophages instead of CD206 as planned.
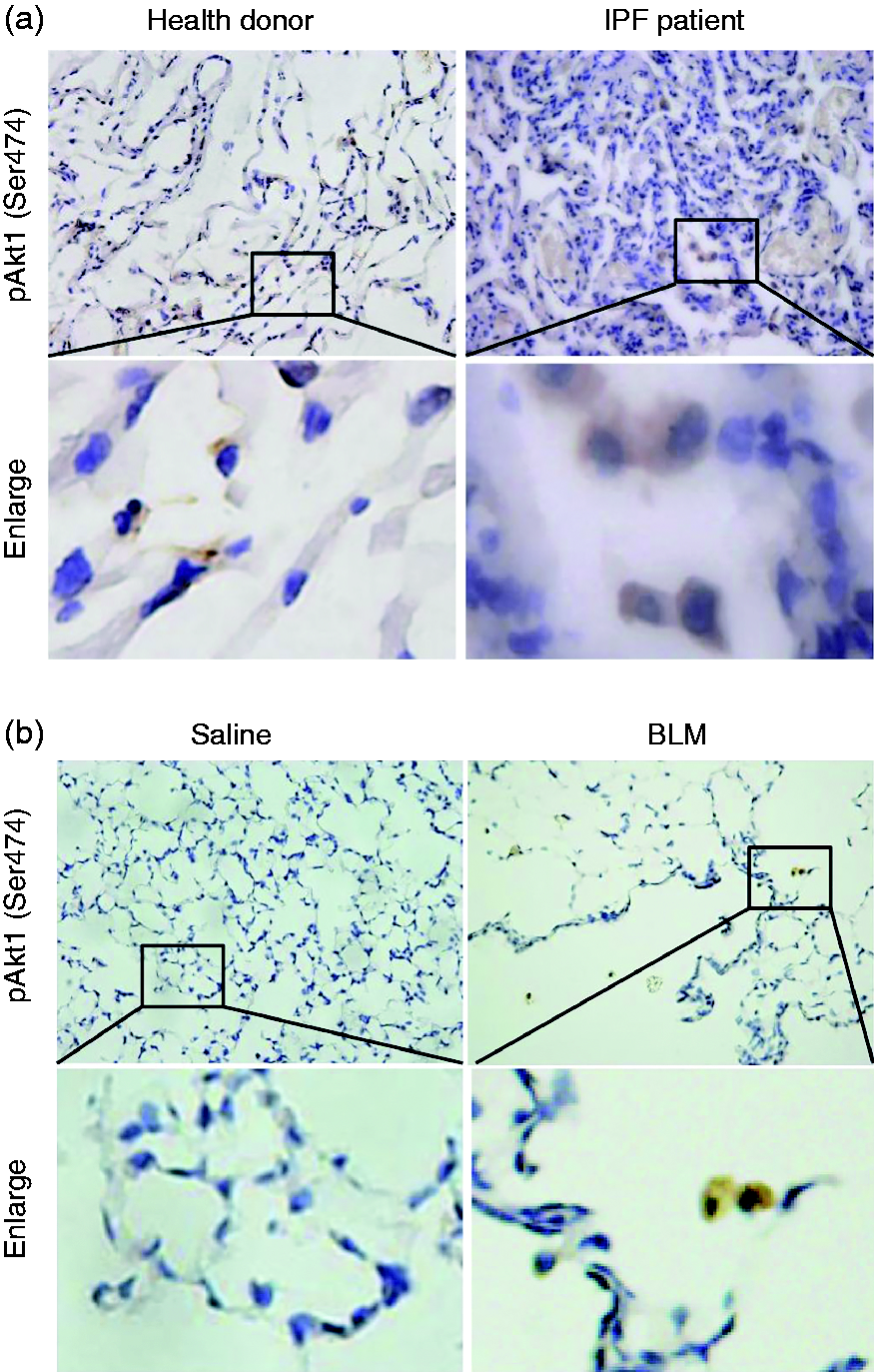
Phospho-Akt1 (Ser473) is up-regulated in lungs from pulmonary fibrosis patients and mice. Phospho-Akt1 (Ser473) in lung tissue from (a) donors or (b) pulmonary fibrosis patients in a bleomycin (BLM)-induced mice model was detected by immunohistochemical staining. Original magnification 400×.
Akt1 conventional knockout reduces pathology of BLM-induced IPF and mortality
To determine whether Akt1 conventional knockout regulates IPF, Akt1+/+ and Akt1–/– mice were given BLM intratracheally (1.4 IU/kg) to induce the development of pulmonary fibrosis. Twenty-one d after intratracheal injection of BLM, histological analysis of the Akt1+/+ lungs showed destruction of the alveolar architecture, massive collagen deposits and accumulation of fibroblasts and myofibroblasts. In comparison, lung sections from Akt1–/– mice showed a well-preserved alveolar structure, fewer fibroblasts and less collagen deposition around the capillary vessels (Figure 2a). Collagen deposition in lung sections by Masson’s trichrome staining showed the pulmonary interstitium from Akt1–/– mice displayed less blue deposition compared to Akt1+/+ mice, suggesting a reduction in collagen accumulation in Akt1-deficient mice (Figure 2b). To assess pulmonary fibrosis further, Ashcroft scores were analysed and were significantly different between Akt1+/+ and Ak1–/– mice (Figure 2c). These data showed that Akt1 deficiency attenuated the change of BLM-induced pathology. We further evaluated collagen deposition by its major component, hydroxyproline content, using a hydroxyproline assay kit. It was found that Akt1 deficiency significantly reduced hydroxyproline content induced by BLM in the lungs. Next, we explored the expression of several markers of IPF. The mRNA level of α-SMA and fibronectin was down-regulated in Akt1-deficient mice (Figure 2d and e). Western blot analysis of the total lung homogenates showed that Akt1 deletion reduced the BLM-induced α-SMA protein level compared to Akt1+/+ mice (Figure 2f and g). Figure 2h showed that hydroxyproline content, a major component of collagen protein, was also significantly lower in Akt1-deficient mice. More importantly, a much improved survival rate was seen in Akt1−/− mice compared to Akt1+/+ mice 21 d after the administration of BLM (Figure 2i). Collectively, these results suggest that Akt1 conventional deficiency ameliorates BLM-induced IPF in mice.
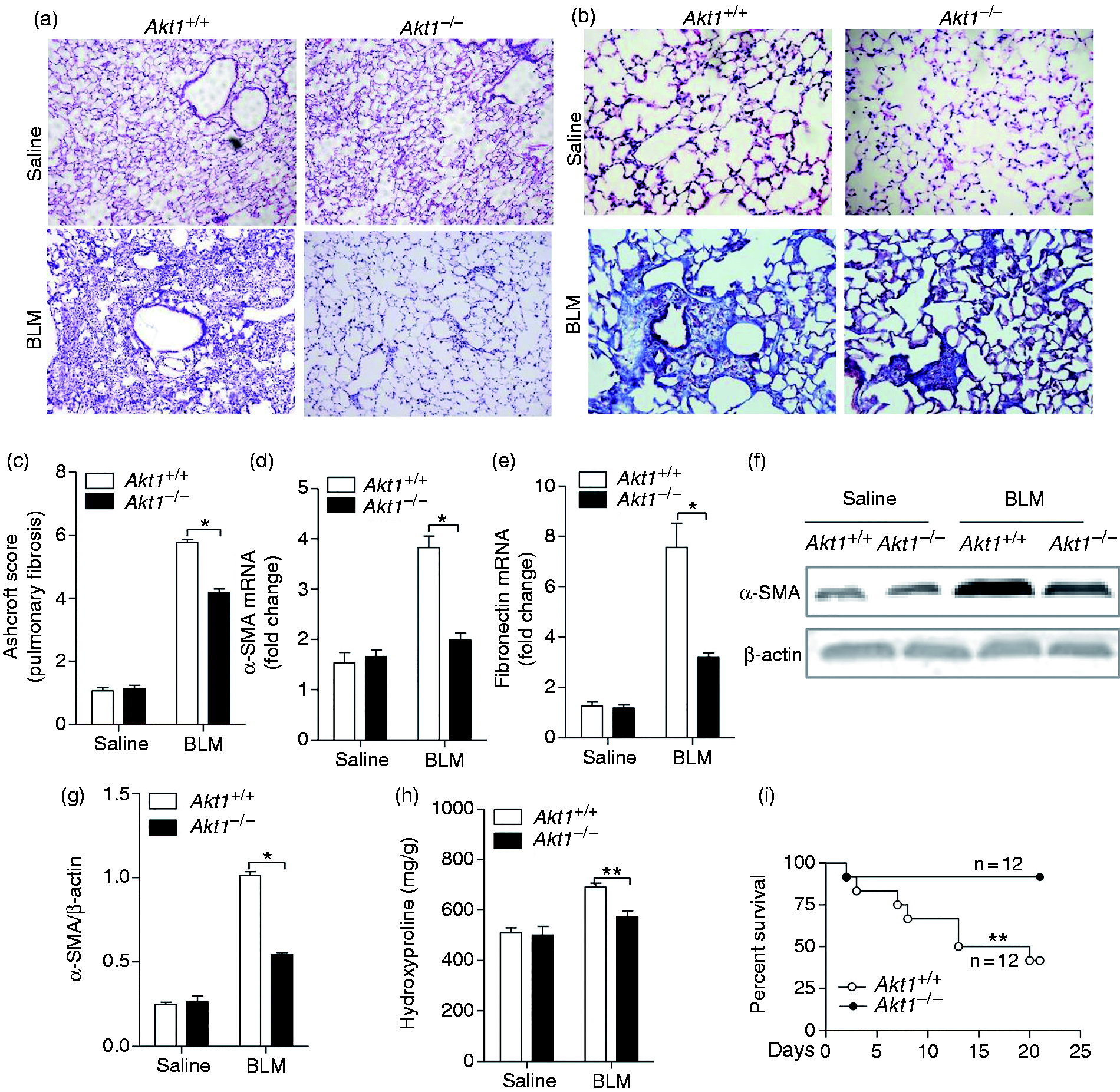
Akt1 conventional deficiency attenuates BLM-induced idiopathic pulmonary fibrosis (IPF) and reduces mortality in mice. (a) Akt1+/+ and Akt1–/– mice were intratracheally injected with saline or BLM (1.4 IU/kg; n = 6–8 mice/group), and lung sections were stained with hematoxylin and eosin (H&E). Original magnification 200×. (b) Masson’s trichrome staining was performed to detect collagen deposition in Akt1+/+ and Akt1–/– mice upon treatment with BLM. Original magnification 400×. (c) Ashcroft score of the lung sections from Akt1+/+ and Akt1–/– mice. (d and e) The expression of alpha smooth-muscle actin (α-SMA) and fibronectin was measured by quantitative RT-PCR in lungs from mice as described. (f) The expression of α-SMA was measured by Western blot in lungs from mice as described. (g) The level of α-SMA was measured by quantitative analysis. (h) Hydroxyproline content was determined using the hydroxyproline assay kit. (i) Age- and mass-matched male Ak1+/+and Akt1–/– mice (n = 2) were challenged with bleomycin (1.5 IU/kg) and monitored for 21 d. The survival curve was recorded. Data are shown as means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to Akt1+/+ mice treated with BLM, two-tailed Student’s t-test.
BLM-Induced lung inflammatory response is reduced in Akt1–/– mice
BLM causes damage to the alveolar epithelial cells, followed by an interstitial inflammatory response within the first wk after its administration, replicating the pathological characteristics of human pulmonary fibrosis. To evaluate whether Akt1 regulated BLM-induced inflammation, we first compared the pathological changes and MPO activities in Akt1+/+ and Akt1–/– mice 3 and 7 d after injection of BLM. As shown in Figure 3a and b, the destruction and inflammatory cell infiltration induced by BLM were significantly reduced in Akt1–/– mice. Next, the lung alveolar permeability was evaluated using the protein concentration and cell counts in BALF. The data showed that conventional Akt1 knockout decreased protein concentration and cell counts of BALF from mice challenged with BLM for 3 and 7 d (Figure 3c and d). Collectively, these data indicate that Akt1 is required for BLM-induced lung inflammation.
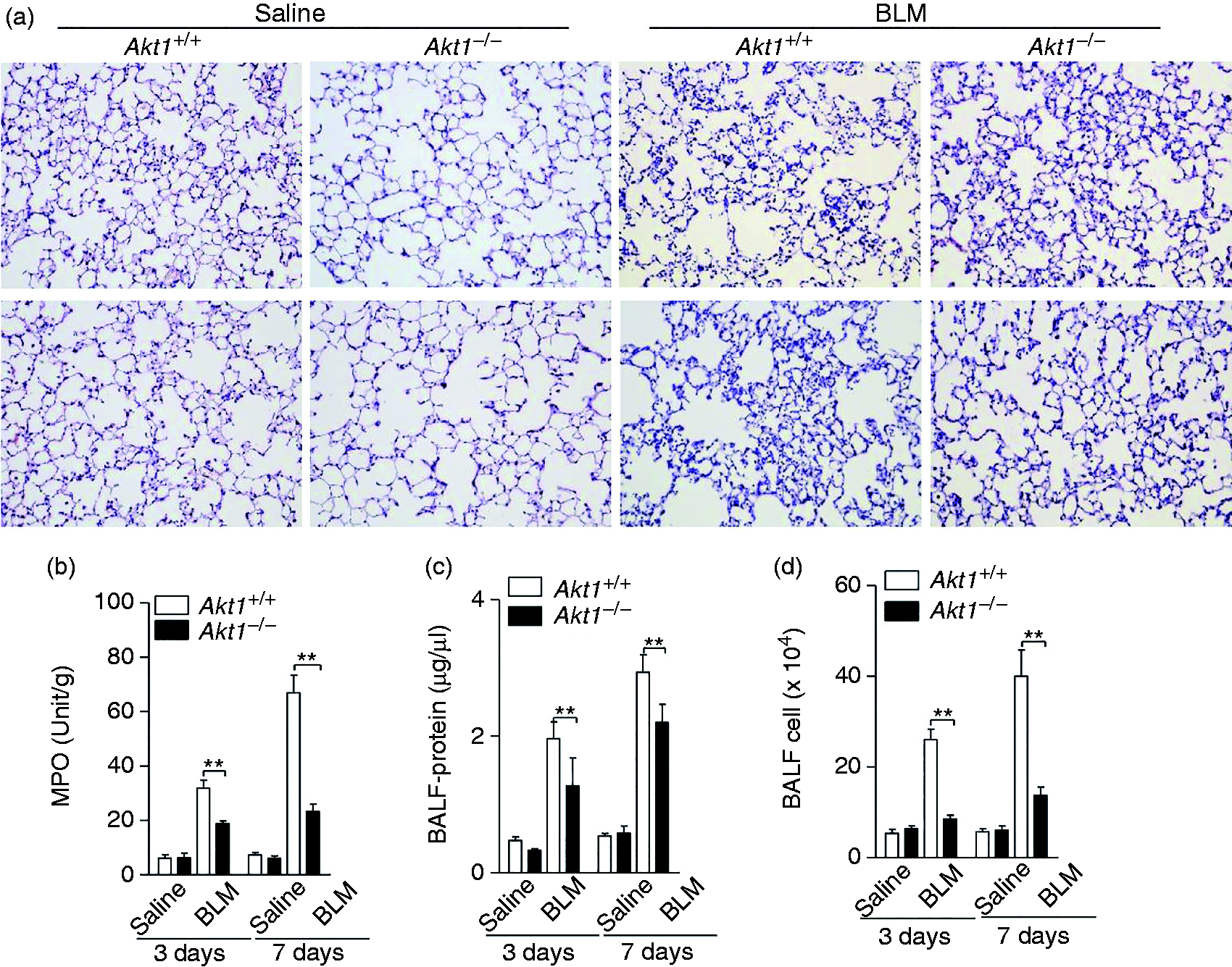
Akt1 conventional deficiency attenuates BLM-induced inflammation. (a) Akt1+/+ and Akt1–/– mice were intratracheally injected with saline or BLM (1.4 IU/kg) for the indicated times (n = 6–8 mice/group). The lung sections were stained with H&E. Original magnification 200×. (b) Myeloperoxidase activity of lung tissues was measured. (c and d) Protein concentration and total cells in bronchoalveolar lavage fluid (BALF) from Akt1+/+ and Akt1–/– mice were quantified. Data are shown as means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to Akt1+/+ mice treated with BLM, two-tailed Student’s t-test.
Akt1 is required for IL-13 production in BLM-induced lungs
It has previously been shown that pro-fibrotic cytokines such as TGF-β, IL-10 and IL-13 play an important role in regulating the pathogenesis of pulmonary fibrosis. We next evaluated the production of these pro-fibrotic cytokines in the lung of Akt1+/+ and Akt1–/– mice on d 3 and 7 after BLM administration. As shown in Figures 4a and b, the production of several pro-fibrotic cytokines TGF-β and IL-10 was not significantly altered in Akt1–/– mice comparing to that from Akt1+/+ mice after BLM treatment. However, notably, the Akt1–/– mice given BLM exhibited a lower mRNA level of IL-13 compared to Akt1+/+ mice treated with BLM (Figure 4c). In order to determine the contribution of macrophages in Akt1-mediated IPF, we evaluated the above gene expression in isolated lung macrophages from IPF models. Similarly, the mRNA level of IL-13 was significantly down-regulated, while the production of TGF-β and IL-10 was not significantly altered in isolated lung macrophages of Akt1–/– mice after BLM treatment (Figure 4d–f).
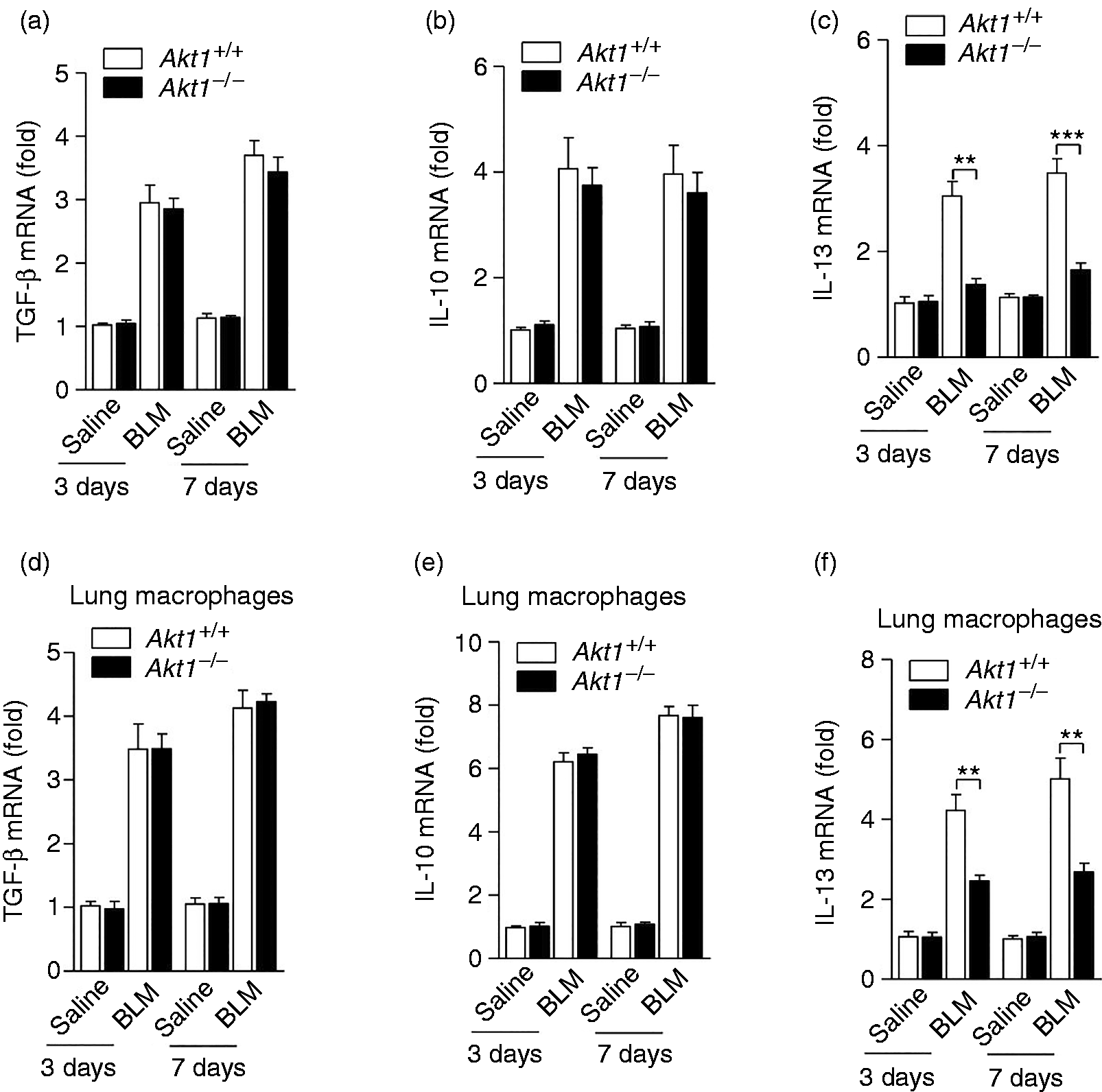
Akt1 is required for IL-13 production in BLM-treated mice. Akt1+/+ and Akt1–/– mice treated with saline or bleomycin (1.4 IU/kg) for 3 and 7 d. Expression of the cytokines (a) TGF-β, (b) IL-10 and (c) IL-13 in the lungs were detected by quantitative RT-PCR; expression of the cytokines (d) TGF-β, (e) IL-10 and (f) IL-13 in isolated lung macrophages were detected by quantitative RT-PCR (n = 6–8 mice/group). Data are shown as means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to treated cells from Akt1+/+ mice, two-tailed Student’s t-test.
Akt1 is activated and required for IL-13 production by IL-33 stimulation in macrophages
During pulmonary fibrosis, IL-33, which is released by injury epithelial and endothelial cells, acts as an endogenous danger signal for the production of several pro-fibrotic cytokines. 29 To explore the regulatory effect of Akt1 on macrophage function upon IL-33 stimulation, we next examined the involvement of Akt1 in the IL-33 signalling pathway. BMDMs isolated from Akt1+/+ mice were treated with IL-33 (10 ng/ml) for 2 h, and an immunoblot assay was performed to detect the activation of Akt and its isoform Akt1. As shown in Figure 5a, there was a significant increase in the phosphorylation of Akt and its isoform Akt1. The quantitative analysis also identified this activation, as shown in Figure 5b and c. Next, we detected the mRNA level of IL-13 and found IL-13 expression significantly decreased from a mean of 19.9-fold in Akt1+/+ macrophages to a mean of 9.04-fold in Akt1–/– macrophages (P = 0.0013) after IL-33 stimulation for 24 h, and from a mean of 13.2-fold in Akt1+/+ macrophages to a mean of 4.44-fold in Akt1–/– macrophages (P = 0.0086) after stimulation for 48 h (Figure 5d). In order to explore how Akt1 promotes IL-13 secretion, we detected the activity of NF-κB and observed decreased phosphorylation of p65 (Ser536; Figure 5e and f). Taken together, our data show that Akt1 regulates production of IL-13 in response to IL-33 stimulation, which may be associated with phosphorylation of p65.
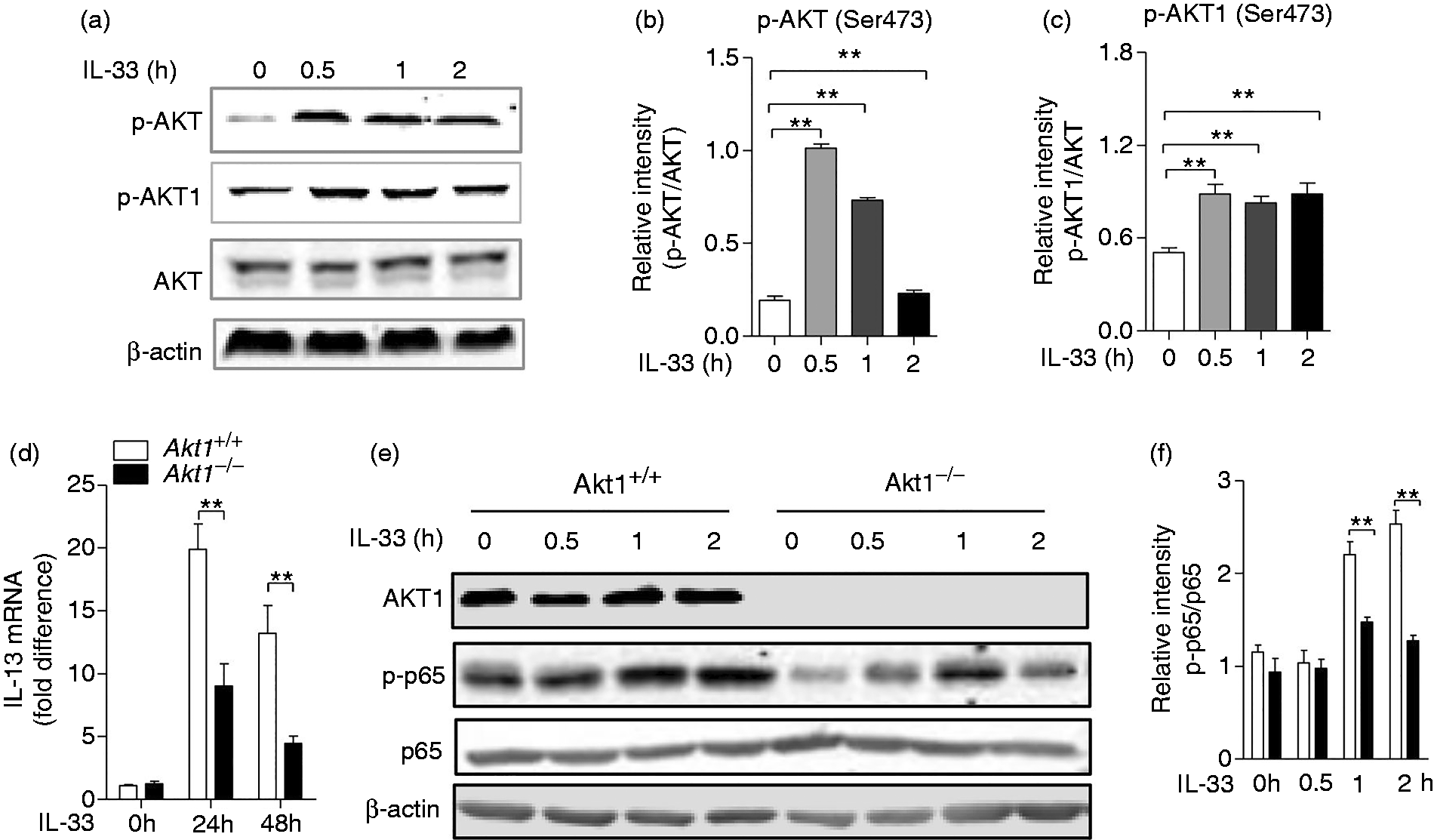
Akt and Akt1 were phosphorylated, and Akt1 is required for IL-13 production in macrophages by IL-33 stimulation. (a) Akt1+/+ bone marrow–derived macrophages (BMDMs) were challenged with IL-33 (10 ng/ml) for 2 h. Phospho-Akt (Ser473), Akt, Phospho-Akt1 (Ser473) and β-actin were measured by Western blot. Quantitative analysis of the relative expression levels of phosphorylated (b) Akt and (c) phosphorylated Akt1 against total AKT using ImageJ software. (d) Akt1+/+ and Akt1–/– BMDMs were treated with IL-33 (10 ng/ml) for 24 and 48 h. The mRNA level of IL-13 was detected by quantitative RT-PCR. (e) The Akt1+/+ and Akt1–/– BMDMs were challenged with IL-33 (10 ng/ml) for 2 h. Phospho-Akt1, Phospho-p65 (Ser536), p65 and β-actin were measured by Western blot. (f) Quantitative analysis of the relative expression levels of phosphorylated p65 against total p65 using ImageJ software. Data are shown as means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to treated cells from Akt1+/+ mice, two-tailed Student’s t-test.
Discussion
IPF is a devastating lung disease involved various cells and a number of mediators.1,30 The present study demonstrated that the phosphorylation of Akt1 was up-regulated in the tissue from pulmonary fibrosis patients, while Akt1 global germ-line deletion attenuated BLM-induced pulmonary fibrosis and inflammation using conventional Akt1 knockout mice in an experimental model of pulmonary fibrosis. Importantly, Akt1 mediated the release of pro-fibrotic cytokine IL-13, and this modulation could be induced under IL-33 treatment, which was consistent with our previous work demonstrating Akt2 has a critical role in pulmonary fibrosis via modulating macrophage function induced by IL-33. 21 Similarly, it has also been confirmed that macrophage-specific Akt1 deletion alleviated BLM-induced IPF through mediating alveolar macrophage apoptosis. 22 However, the correlation of Akt1, macrophage derived pro-fibrotic cytokines and IPF has still to be elucidated. The current study illustrated that Akt and its isoform Akt1 were both phosphorylated in macrophages induced by IL-33, with lower IL-13 induction in the Akt1 knockout group. This indicated that by regulating macrophage function activated by IL-33, Akt1 plays a key role in pulmonary fibrosis, which could be attributed to the critical roles of macrophages played via complex and flexible signalling pathways in the development of fibrosis.3,31
Akt, comprised of three closely related isoforms Akt1, Akt2 and Akt3, was identified as exhibiting a series of biological effects, including cell proliferation, apoptosis, survival and metabolism.32,33 Recent studies reported that Akt components play distinct and substrate-specific roles in macrophage subtypes and inflammatory disease by producing inflammatory cytokines.34–36 Akt2 ablation promotes alternatively activated macrophages and protects from LPS-induced endotoxin shock and dextran sulfate sodium (DSS)-induced colitis. Previous work has shown that Akt2 regulated IPF via modulating IL-13 and TGF-β of macrophages induced by lung injury in vivo and IL-33 in vitro. 37 Recent studies have shown that hyper-activation of Akt1 induced focal fibrosis similar to TGF-β-induced fibrosis. 38 Akt1, highly expressed on various lung cells, was reported to exhibit opposite effect of Akt2 on macrophage polarisation and to be more sensitive to LPS-induced endotoxin shock and DSS-induced colitis. However, with regard to the inflammatory disease and pulmonary fibrosis, Akt1 was identified as playing negative roles in their development. Hyper-activation of Akt1 induced focal fibrosis similar to TGF-β-induced fibrosis. 38 Macrophage-specific deletion of Akt1 relieves pulmonary fibrosis via mediating macrophage apoptosis. 22 The role of Akt1 in macrophage-derived pro-fibrotic cytokines during the IPF process remains to be elucidated.
In lung tissue, IL-33 is constitutively expressed in endothelial cells and epithelial cells in response to injury. The IL-33 level was elevated in the lungs from IPF patients and BLM-injury mouse models.39,40 IL-33 was identified as inducing the expression of pro-fibrotic cytokine IL-13 in a variety of cells, including macrophages.41,42 Extracellular IL-33 binds to ST2 and activates the heterodimeric receptor complex ST2/IL-1RAcP on the plasma membrane, followed by the sequential activation of MyD88, IRAK4, TRAF6, TAK1 and MAPK. 43 Then, the nuclear transcription factors are activated to mediate the production of IL-13. NF-κB is one of these important nuclear transcription factors. It is well established that the activation of NF-κB (P65) results in IL-33-induced IL-13 production in various cell types, including macrophages.43–45
Our results showed that IL-33 activated Akt and its substrate Akt1. Akt1 may play a role in BLM-induced IPF through the IL-33 signalling pathway. Using global Akt1 knockout mice, we found Akt1 deficiency resulted in lower collagen deposition and fibrogenesis. IL-13 has emerged as a central mediator of pulmonary fibrosis. Overexpression of IL-13 leads to pulmonary fibrosis, and our data show that Akt1 deficiency reduces IL-13 release in the lung after BLM treatment for 3 and 7 d. Consistently, the level of IL-13 was down-regulated in Akt1–/– macrophages treated with IL-33. Meanwhile, the increased activity of p65 by IL-33 stimulation was down-regulated in Akt1-deficient macrophages. All these data show that Akt1 may also regulate IPF through macrophage-derived IL-13, that the function of macrophage in Akt1-mediating IPF is vital and that P65 may be associated with the effect of Akt1 on macrophages. Interestingly, we found the phosphorylation of Akt1 is up-regulated in the lungs from IPF patients, which is similar to the phenomenon of Akt2.
Akt kinase isoforms are similar in structure, although studies over the last decade using various transgenic and knockout mice have shown that Akt isoforms have distinct functions and distribution. They are partly redundant, and both Akt1 and Akt2 are ubiquitously and highly expressed in the lung.46,47 Akt1 and Akt2 may compensate the function for each other if they share the common signalling pathway, while the current study uncovers a novel mechanism of Akt1 in regulating IPF. Akt1 and Akt2 could act as substitutes for each other when blocking one, which should be taken into account when exploring either as a therapeutic target for IPF.
In conclusion, the present study suggests that Akt1 modulates pulmonary fibrosis through up-regulating the production of the pro-fibrotic cytokine IL-13 in macrophages. The convergence of Akt1 in regulating IPF via modulating IL-13 production of macrophage as demonstrated here not only contributes to our understanding of common and distinct Akt isoforms, but also has important implications in exploring potential therapeutic strategies for IPF. Furthermore, the mechanism of Akt1 in regulating IL-13 merits further investigation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by National Natural Science Foundation of China (81800065 and 81773741), Natural Science Foundation of Jiangsu Province (BK20180616) and Youth Natural Science Foundation of Jiangnan University (JUSRP11864).