
Abstract
Impaired Paneth cell expression of antimicrobial protein (AMP) lysozyme is found in patients with Crohn’s disease with the autophagy gene ATG16L1 risk allele, in mice with mutations in autophagy genes Atg16L1, Atg5 and Atg7, and in Irgm1 knockout mice. Defective autophagy is also associated with expansion of resident Gram-negative bacteria in the intestinal lumen. These findings suggest that autophagy may control extracellular resident microbes by governing expression of lysozyme. To test the hypothesis that autophagy may have a defensive role in host response to resident extracellular microbes, we investigated the relationship between gut microbes, autophagy, and lysozyme. RAW 264.7 macrophages were treated with fecal slurry (FS), representing the resident microbial community; lipopolysaccharide (LPS); or butyrate, representing microbial products; or a representative resident Gram-negative bacterium Desulfovibrio vulgaris (DSV). FS, LPS, and DSV inhibited lysozyme expression, whereas butyrate had no effect. Induction of autophagy by rapamycin countered this inhibition, whereas silencing of the autophagy gene Irgm1 exacerbated the inhibitory effects of LPS on lysozyme expression. LPS also inhibited lysozyme activity against DSV and autophagy reversed this effect. Our results provide a novel insight into an interaction between gut bacteria, autophagy and AMP whereby autophagy may defend the host by countering the suppression of antimicrobial protein by Gram-negative bacteria.
Introduction
Autophagy has been well characterized for its function in eliminating intracellular pathogens. However, its potential role in controlling extracellular, non-pathogenic resident gut microbes remains unexplored. A recent study provided clues suggesting this function of autophagy. Crohn’s disease (CD) patients homozygous for the ATG16L1 T300A risk allele were found to have an increased number of Gram-negative resident gut microbes in the intestinal lumen belonging to the families Enterobacteriaceae, Bacteroidaceae and Fusobacteriaceae. 1 The underlying mechanism of this association with the mutated autophagy gene remains unknown. Another study on CD patients homozygous for the ATG16L1 T300A risk allele found that there was an abnormal distribution of antimicrobial protein lysozyme in intestinal Paneth cells. 2 This finding was further validated in mouse models of defective components of autophagy machinery, such at Atg5, Atg16l1, Atg7 and Irgm1.3,4
Antimicrobial peptides/proteins (AMPs) are ancient immune defense mechanisms that are found in all organisms. AMPs have potent microbial killing properties against wide spectrum of organisms, including bacteria, viruses, fungi, and protozoa. AMPs are expressed both broadly and in specialized cells and occur as inducible or constitutively expressed molecules. Some examples of AMPs include lysozyme, phospholipase A2, cathelicidins, α-defensins (e.g. HD5 and HD6), and β-defensins (e.g. HBD-1 and HBD-2). AMPs not only protect the host against invading pathogens, but also shape the composition of resident commensal gut microbes. 5 Defective AMP production was found to be associated with alteration of gut microbial community. 6
These findings suggest that autophagy may also have a role beyond the defense against intracellular pathogens. Could autophagy also control the extracellular resident gut microbial population via secretion of AMPs such as lysozyme? In this study, we tested the hypothesis that autophagy may have a defensive role in the host response to resident extracellular microbes by testing the relationship between gut microbes, autophagy, and lysozyme in vitro in macrophages.
Materials and methods
Cell culture and treatments
RAW264.7 cells were purchased from and grown according to ATCC methods, in DMEM supplemented with 10% FBS. No antibiotics or any other antimicrobial reagents were added to the culture medium, as they were found to interfere with the LPS inhibitory effects on lysozyme expression. Cells were treated for 3 h with filtered fecal slurry (in PBS) obtained from euthanized mice, or with LPS (L4391; Sigma, St. Louis, MO, USA), sodium butyrate (B5887; Sigma) or Desulfovibrio vulgaris (DSV) for 24 h. Polymyxin (P4932; Sigma) was added to a final concentration of 0.1 mg/ml 1 h before the LPS challenge. Rapamycin (R8781; Sigma) at 100 nM was added to the cells 1 h prior to challenge with test reagents. DMSO-treated cells (volume equivalent to what was used for 100 nM rapamycin) were used as controls. Rapamycin was also added to the cells 24 h after LPS challenge. In this case, rapamycin was added for 24 h in the continued presence of LPS. Cells were harvested and processed for preparation of protein and RNA samples.
DSV culture
The sulfate-reducing bacterium, DSV Hildenborough NCBI 8303, was grown on a lactate–sulfate medium according to previously published methods. 7
siRNA transfection
siRNA against mouse Irgm1 was purchased from Dharmacon (Lafayette, CO, USA). Thirty-five nM siRNA was used for transfection of RAW cells. Transfection was carried out with Lipofectamine 2000 (52758; Thermo Fisher Scientific, Waltham, MA, USA) using manufacturer’s protocol. At 24 h post-transfection, cells were trypsinized and plated into six-well plates. After an additional 24 h, cells were treated with the test reagents. Irgm1 knock down was confirmed with Western blotting using Irgm1 Ab (sc11075; Santa Cruz, CA, USA).
Western blot
RAW cells were lysed in Lysis buffer (87787; Thermo Fisher Scientific) containing protease and phosphatase inhibitors (1861281; Thermo Fisher Scientific). Fifty µg protein samples were run on SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were blocked in 5% milk for 30 min followed by overnight (∼18 h) incubation in lysozyme Ab (ab108508; Abcam, Cambridge, UK), actin Ab (4970; Cell Signaling, Danvers, MA, USA) or LC3 Ab (27755; Cell Signaling). Actin and LC3 Abs were diluted according to the manufacturer (1:1000 in 5% BSA). Lysozyme Ab was diluted 1:5000 in 5% milk. All the dilutions were made in wash buffer PBS–0.1% Tween 20. Blots were incubated with secondary Abs (7074; Cell Signaling) at room temperature (∼20°C) for 1 h (dilution of 1:2000). Blots were developed using enhanced chemiluminescence (32106; Thermo Fisher Scientific).
Quantitative PCR
RNA (RNeasy Mini Kit, 74106; Qiagen, Hilden, Germany) was extracted from cells and cDNA synthesis was carried out using cDNA kit (18080-051; Thermo Fisher Scientific). Real-time quantitative PCR (qPCR) was performed using SYBR green (204145; Qiagen). The primer sequences were as follows: 18s F: GTAACCCGTTGAACCCCAT; 18 s R: CCATCCAATCGGTAGTAGCG; Lyz F: GGATCAATTGCAGTGCTCTG; Lyz R: CAGTTCCGAATATACTGGGAC. qPCR conditions were as follows: 95°C for 15 min for 40 cycles; then 30 s at 95°C, 30 s at 51°C and 30 s at 72°C; and for the melting curve: 95°C for 15 s, 60°C for 30 s, 95°C for 15 s
Filtered mouse fecal slurry
The luminal contents of the cecum and colon were collected from mice and re-suspended in PBS. The slurry was filtered through glass wool followed by filtration through 0.2 -µm filters. The filtrate was then added to RAW cells for 3 h.
Lysozyme activity assay
The protocol for lysozyme activity of the cells against Micrococcus lysodeikticus (M3770; Sigma) was adopted from a published protocol. 8 Enzyme activity against DVS in the culture supernatant of RAW cells was analyzed as described. 9
For M. lysodeikticus
Briefly, cells were lysed and protein concentration was normalized to 1 mg/ml. Micrococcus lysodeikticus was prepared (2 mg/ml in PBS, pH 7.0) and further diluted to obtain ∼0.7 OD450. Two hundred µl of this solution was mixed with 100 µl protein sample in a 96-well plate in triplicates and OD450 was analyzed at 90 min.
For DSV
DSV culture was centrifuged and re-suspended in 0.1 M EDTA in 0.05 M Tris pH 9.0 and incubated in ice for 10 min. Bacteria were washed in 0.05 M Tris pH7.0 and re-suspended in the same buffer. The bacterial solution was then diluted to the OD600 of ∼0.8. One ml of this solution was added to 100 µl of the culture supernatant (prepared by concentrating 2 ml of culture supernatant using Amicon filters to a volume of 100 µl). OD600 was analyzed at 60 min.
Lactate Dehydrogenase (LDH) cytotoxicity assay
LDH activity was determined using a commercially available kit (8895; Thermo Fisher Scientific) based on the manufacturer’s protocol.
Lysozyme detection in the culture supernatant
Culture supernatants (for extracellular lysozyme), as well as the cell lysates (for cellular lysozyme), were precipitated with nine volumes of ice-cold ethanol and stored at –20° C for 1 h. The samples were centrifuged at 13,000 g for 15 min at 4°C and the supernatant discarded. The pellets were air dried and re-suspended in PBS. Protein concentration was measured with Bradford reagent and 50 µg protein from the supernatant or the lysate was analyzed by Western blotting to detect lysozyme.
Statistical analysis
All graphs were generated using Graph Pad Prism 5 (GraphPad, La Jolla, CA, USA). For qPCR data analysis, we compared differences between the treatments groups using a two-tailed t-test. P-Values < 0.05 were considered significant.
Results
Resident gut microbes, LPS and resident gut bacterium DSV inhibited lysozyme expression
To test directly the relationship between gut microbial products, autophagy and AMP, we carried out in vitro studies using RAW 264.7 macrophages. In the gut, lysozyme is produced mainly by macrophages and Paneth cells. While Paneth cells are found primarily in the ileum, macrophages are present throughout the length of the gut. 10
We first tested the effect of filtered mouse fecal slurry (FS), comprised of metabolites and other gut microbial products, as a surrogate for gut microbes on lysozyme expression in RAW cells. We found that FS had an inhibitory effect on lysozyme expression (Figure 1a). We then tested the effect of bacterial endotoxin LPS, one of the most abundant bacterial products found in feces.
11
We found that purified LPS also inhibited lysozyme expression, an effect similar to FS (Figure 1a). Quantification of Western blot revealed a significant difference in lysozyme expression in control (1.13 ± 0.05) vs. FS (0.32 ± 0.14; P < 0.01) and in control (0.99 ± 0.04) vs. LPS-treated cells (0.10 ± 0.03; P < 0.001). This is consistent with previously published studies on the inhibition of lysozyme expression by LPS in macrophages.12,13 FS and LPS also inhibited lysozyme expression at the mRNA level (Figure 1b). Compared with control cells (1.01 ± 0.10), a significant decrease was observed in lysozyme expression in FS-treated (0.62 ± 0.02; P < 0.05) and LPS-treated (0.43 ± 0.14; P < 0.05) cells. LPS induced an increase in IL-1β mRNA level in our experiments, confirming its expected pro-inflammatory role (data not shown).
FS and LPS down-regulated lysozyme (a) RAW 264.7 cells grown on a six-well plate were treated with freshly prepared FS (200 µl) for 3 h and LPS (100 ng/ml) for 24 h and cells were processed for immunoblotting with lysozyme antibody. The ratio of lysozyme to actin was quantified using ImageJ software (NIH, Bethesda, MD, USA). Graph represents mean ± SEM (n = 3); **P < 0.05, ***P < 0.01 (two-tailed t-test). (b) RNA was isolated from the control or the FS- or LPS-treated cells and lysozyme transcription levels were analyzed with qPCR. Results are shown as mean ± SEM; *P < 0.05 (two-tailed t-test). (c) Cells were treated with different concentrations of NaBu for 24 h. Cells were further incubated in the presence or absence of LPS (100 ng/ml) for 24 h and analyzed by Western blotting for lysozyme expression (d). Cells were treated with polymyxin B (0.1 mg/ml) for 1 h prior to LPS challenge for 24 h and processed for Western blotting.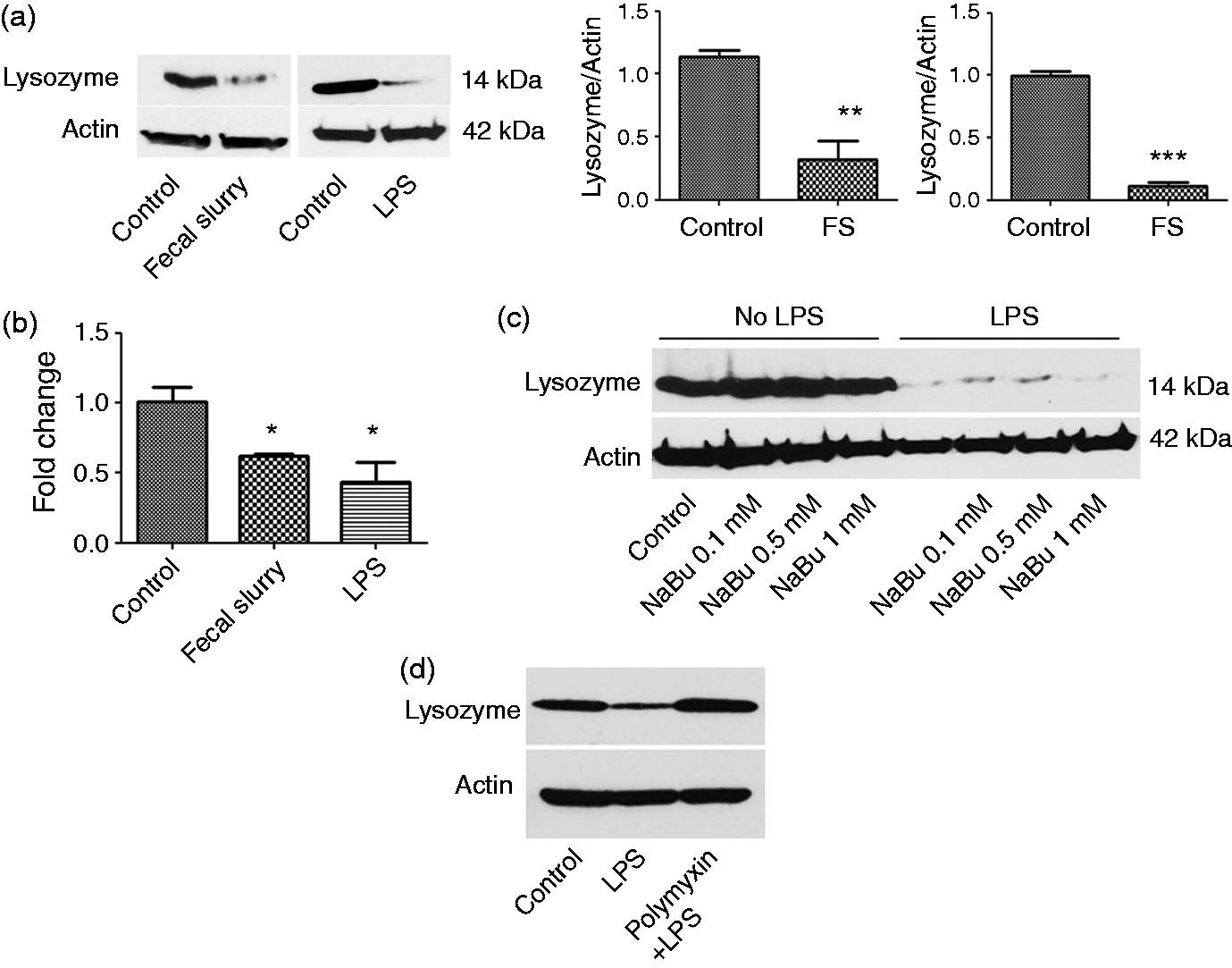
To test whether or not the inhibitory effect of LPS is specific we next tested the effect of butyrate, a Gram-positive bacterial product, on lysozyme expression. Butyrate, a microbe-derived short-chain fatty acid, is a nutrient vital for the host and has anti-inflammatory properties. 14 We found that treatment with sodium butyrate (NaBu) had no effect on lysozyme production (Figure 1c). Moreover, pre-incubation of cells with butyrate did not confer protection against inhibitory effect of LPS on lysozyme. Thus, inhibition of lysozyme may be specific to certain bacterial products such as LPS produced by Gram-negative bacteria.
We further tested the effects of LPS on lysozyme expression in the presence of polymyxin B, a LPS neutralizing agent. We found that in the presence of polymyxin B there was no inhibition of lysozyme by LPS (Figure 1d).
Autophagy counters the inhibitory effect of LPS and DSV on lysozyme
We next investigated the effect of autophagy on the inhibition of lysozyme by LPS. Using siRNA, we silenced the autophagy gene for the protein Irgm1.
15
Suppression of Irgm1 exacerbated the inhibitory effect of LPS on lysozyme (Figure 2a). A significant difference was observed in protein levels of lysozyme in the presence of LPS in scrambled (scr) (0.55 ± 0.11) vs. Irgm1 knockdown cells (siIrgm1) (0.19 ± 0.01; P < 0.05) (Figure 2b). Conversely, induction of autophagy with rapamycin protected against the effects of LPS on lysozyme when cells were treated with rapamycin prior to challenge with LPS (Figure 2c, d). While a significant difference was observed in lysozyme expression in control cells (1.0 ± 0.19) compared with LPS-treated cells (LPS) (0.27 ± 0.10; P < 0.05), no difference was observed between control (1.0 ± 0.19) and rapamycin plus LPS-treated cells (Rapa + LPS) (0.65 ± 0.21; P > 0.05). This was also true at the mRNA level where a significant difference was observed between LPS-treated (0.24 ± 0.07) without pre-treatment vs. cells pre-treated with rapamycin (1.13 ± 0.19; P < 0.05) (Figure 2e). Activation of autophagy by rapamycin was confirmed by showing an increase in expression of the autophagic marker LC3II. We observed a slight increase in autophagy by LPS, which is in line with the other studies (Figure 2c).16,17 However, a mild induction of autophagy by LPS alone was not sufficient to overcome the inhibitory effects of LPS on lysozyme.
Autophagy overcomes inhibitory effect of LPS on lysozyme. (a) Cells were transfected with control or Irgm1 siRNA for 48 h and then incubated with different LPS concentrations for 24 h and analyzed for lysozyme by immunoblotting. Knockdown was confirmed with Irgm1 Ab. (b) Quantification of lysozyme protein in scrambled vs. siIrgm1 knockdowns in the absence of presence of LPS. Graph represents mean ± SEM (n = 3); *P < 0.05 (two-tailed t-test). (c) Cells were incubated with or without rapamycin at different concentrations for 1 h and challenged with LPS (0.1 or 0.5 µg/ml) for 24 h in the presence or absence of the drug. (d) RAW cells were pre-incubated with or without rapamycin before LPS treatment. Lysozyme/actin ratio was quantified using ImageJ. Data represents mean ± SEM (n = 3); *P < 0.05, †P > 0.05 (two-tailed t-test). Fold change differences compared with control were reported for all quantifications. Lysozyme was assessed at mRNA level. (e) RAW cells were pre-incubated with or without rapamycin and further incubated with LPS. Lysozyme was assessed at mRNA level. Data represent mean ± SEM (n = 3); *P < 0.05, ***P > 0.05 (two-tailed t-test). (f) Cells were treated with LPS (100 ng/ml) for 24 h followed by treatment with different doses of rapamycin (nM) in the presence of LPS for another 24 h. Cells were then processed for Western blotting.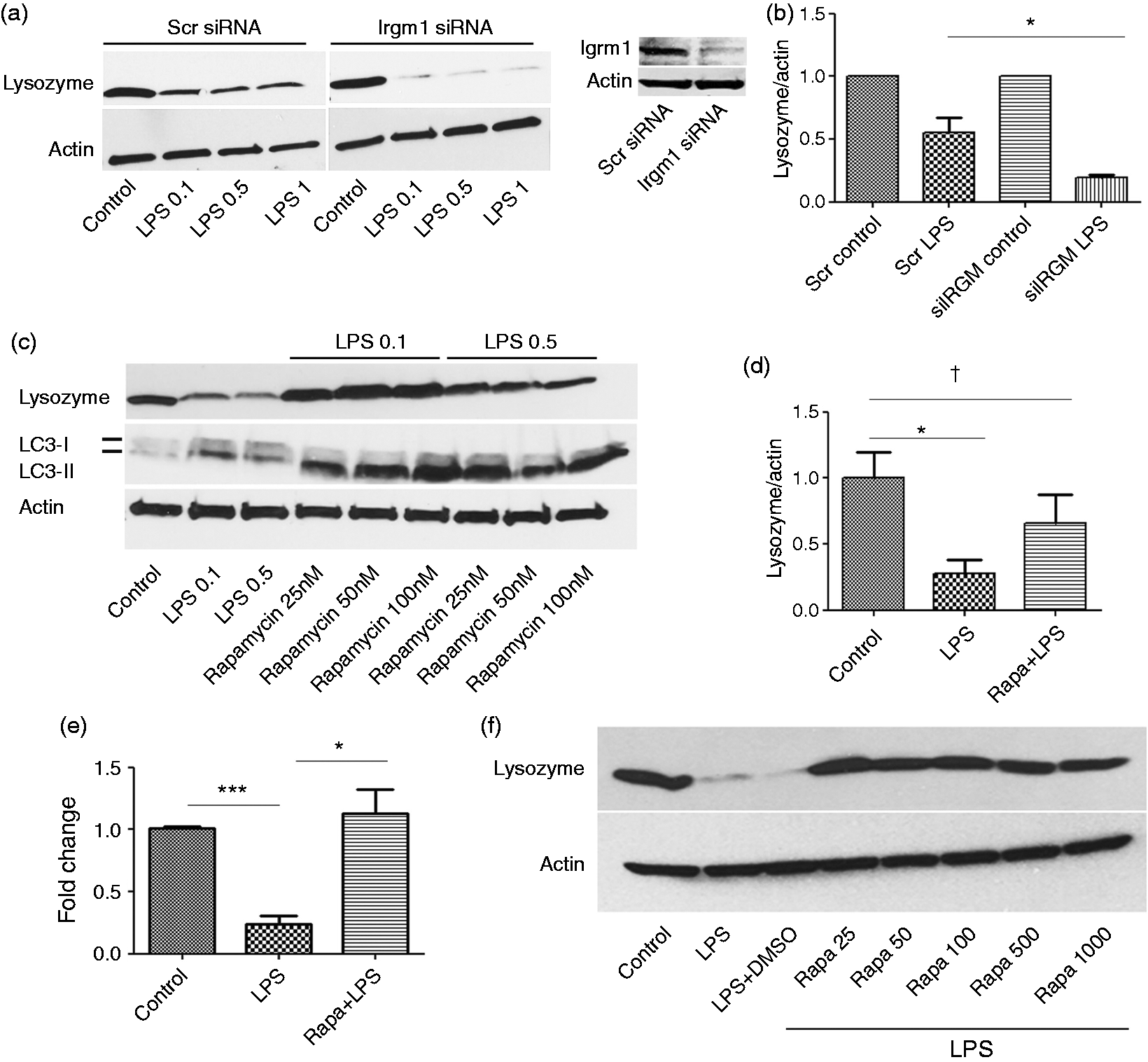
We next investigated the effect on lysozyme expression when rapamycin was added after LPS treatment. Cells were first treated with LPS (100 ng/ml) for 24 h and subsequently treated with various doses of rapamycin for 24 h in the continued presence of LPS. We found that rapamycin reversed the inhibitory effects of LPS on lysozyme expression (Figure 2f). Thus, rapamycin not only conferred protection against subsequent challenge with LPS, but also reversed the inhibitory effects on lysozyme in cells previously treated with LPS.
To test the direct effect of a Gram-negative resident microbe, we also tested the effect of DSV on lysozyme expression in RAW cells. DSV is an LPS-producing Gram-negative bacterium that belongs to a group of sulfate-reducing bacteria which are found in healthy subjects but may be increased in number in patients with inflammatory bowel diseases such as ulcerative colitis.18,19 DSV inhibited lysozyme protein expression comparable with LPS (Figure 3a), and induction of autophagy by rapamycin (Rapa + DSV) overcame this inhibitory effect. We found the DSV also inhibited the lysozyme gene expression (control 1 ± 0 vs. DSV 0.30 ± 0.07; P < 0.05) in RAW cells and these effects could be reversed by rapamycin (DSV 0.30 ± 0.07 vs. Rapa + DSV 1.04 ± 0.17; P < 0.05) (Figure 3b).
Autophagy overcomes inhibitory effect DSV on lysozyme. (a) Cells were treated with DMSO or rapamycin (100 nm) 1 h prior to adding DSV for 24 h. (b) Lysozyme mRNA levels were assessed in the absence or presence of rapamycin prior to treatment with DSV. Data represent mean ± SEM (n = 3); *P < 0.05, †P > 0.05 (two-tailed t-test).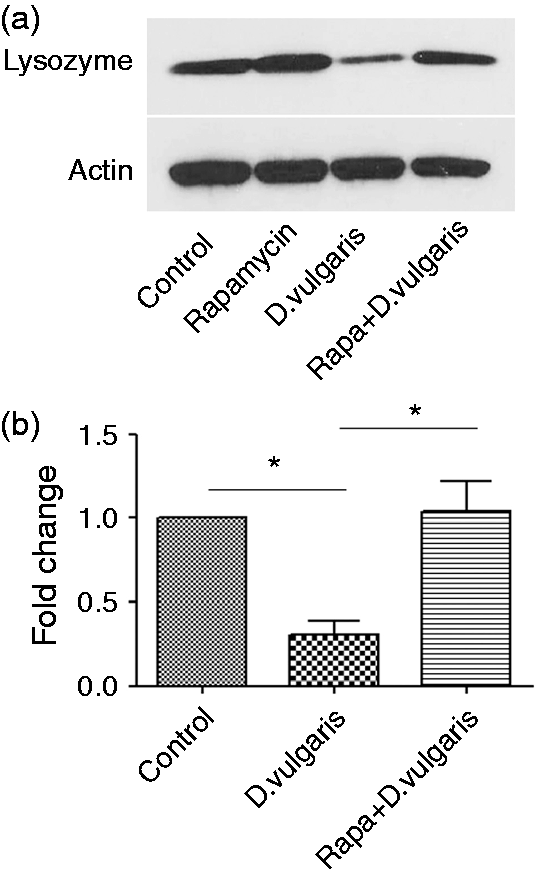
We next tested the effect of LPS and rapamycin treatment on cell viability using lactate dehydrogenase activity assay (LDH) (Figure 4a). LDH is released into the culture medium when the plasma membrane is damaged. The amount of the enzyme released is measured by it enzymatic activity. The culture supernatants were collected and analyzed for LDH activity. The supernatant:pellet ratio of LDH activity was calculated and values were plotted as fold change difference vs. control. We observed a small increase in LDH activity with LPS treatment (1.38 ± 0.04) vs. control (1.00 ± 0.09; P < 0.05). Prior treatment with rapamycin caused a reduction in LDH activity (1.19 ± 0.041) vs. LPS (1.38 ± 0.04; P < 0.05) (Figure 4a).
Effect of rapamycin and LPS on lysozyme secretion and activity. (a) RAW 264.7 cells were tested for viability with LDH cytotoxicity assay kit in response to treatment with LPS in the absence of presence of rapamycin. Values are represented as the ratio of supernatant to pellet activity and are normalized against control. (b) Proteins in culture supernatants and cell lysates were precipitated using ethanol as described. Fifty µg protein was loaded on SDS-PAGE for Western blot analysis for lysozyme detection. (c, d) Densitometric analysis of Western blot using ImageJ for (c) cellular lysozyme, as a ratio of lysozyme to actin and (d) extracellular lysozyme. Values were normalized against controls in both cases. (e) Cells were pre-treated with DMSO or rapamycin and further incubated with LPS (100 ng/ml) for 24 h. Cells were lysed and lysozyme activity was measured using M. lysodeikticus as substrate. (f) Cells were treated the same as before, but the culture supernatant was collected after treatment of the cells and checked for the lytic activity of lysozyme on DSV. Results represented as mean ± SEM. Data are from three different experiments. *P < 0.05; **P < 0.01 (two-tailed t-test).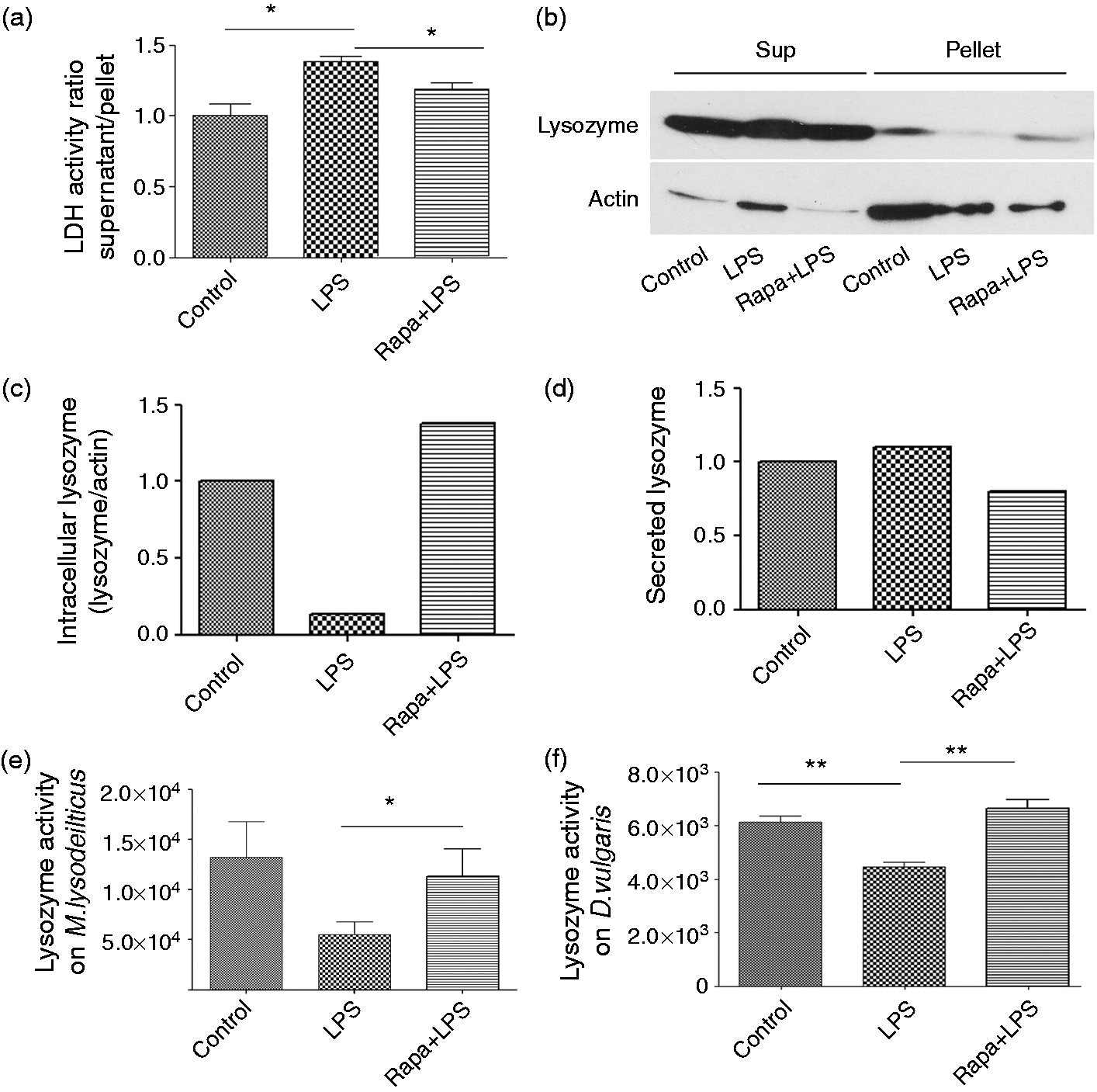
To check whether the observed inhibition of lysozyme by LPS was due to increased secretion of lysozyme, we analyzed the levels of extracellular lysozyme in the culture supernatant following treatments (Figure 4b). While the levels of lysozyme in cell lysates yielded expected results (Figure 4c), the levels of secreted lysozyme appeared comparable in control, LPS- and rapamycin+ LPS-treated cells (Figure 4d). These results argue away from the possibility that the observed decrease in cellular level of lysozyme in LPS treated cells was due to increased secretion of lysozyme.
Given our findings, we further tested if decreased expression of lysozyme in response to LPS corresponded to decreased biological activity of lysozyme and if autophagy protected against this effect. We found that LPS treatment caused a decrease in the antimicrobial activity of lysozyme against the standard target M. lysodeikticus (control 13263.7 ± 3527 vs. LPS 5505 ± 1297) and pre-treatment with rapamycin countered this effect (Rapa + LPS 11343 ± 2751 vs. LPS 5505 ± 1297; P < 0.05) (Figure 4e).
The lytic effect of pure lysozyme on DSV has been previously documented. 9 When we tested the lytic activity of culture supernatants of RAW cells on DSV, a significant inhibitory effect in enzyme activity was observed between the control (6135.4 ± 239.2) compared with LPS-treated supernatant (4442.19 ± 182.7; P < 0.01) (Figure 4f). Pre-incubation of cells with rapamycin prior to LPS challenge recovered lytic activity in the LPS-treated supernatant (6649.83 ± 332.1; P < 0.01).
Discussion
In this study, we investigated the relationship between gut microbial products, autophagy and lysozyme to test the hypothesis that autophagy may have a role in the host response to resident extracellular microbes. For this purpose, we selected RAW 264.7 macrophages. Macrophages play a key role in the host innate immune response. 20 These cells are located in the strategic boundary between the luminal content and the host and contribute to innate immunity via phagocytosis and secretion of lysozyme and anti- and pro-inflammatory cytokines. 21 Macrophages in the gut protect against invading pathogens while maintaining tolerance to the resident commensal microbes. In the intestine, lysozyme is also secreted by Paneth cells. However, while Paneth cells are found primarily in the ileum, macrophages are present throughout the length of the gut. 10 These properties and functions of macrophage make them suitable candidates to study host response to microbes and microbial products.
We found that FS suppressed lysozyme expression suggesting that the predominant effect of resident gut microbes is inhibition of host lysozyme expression. To further break down the components in FS, we tested the effect of LPS, a Gram-negative bacterial product and butyrate, a Gram-positive product on lysozyme expression. We found that LPS but not butyrate inhibited lysozyme expression. This is consistent with previous studies showing inhibition of lysozyme by LPS in mouse macrophages,12,13 but divergent from LPS obtained from Salmonella enterica sv. Typhimurium, which stimulated lysozyme production in chicken myelomonocytic cells and in Atlantic salmon macrophages.22,23 LPS derived from Escherichia coli 0127:B8 did not have any effect on lysozyme activity in RAW 264.7 cells. 8 Thus, LPS has been shown to be both inhibitory and stimulatory for lysozyme production with specific effect of LPS depending on the source, concentrations and the length of treatment and the targeted cells. The inhibitory effect of LPS on lysozyme in our study was not universal to bacterial products as we did not observe the same phenomena with butyrate, another gut microbial metabolite.
We tested the effect of a representative LPS-producing Gram-negative resident gut bacterium, DSV, which is not an intracellular pathogen. We found that DSV inhibited lysozyme expression, possibly owing to its LPS effects. This result demonstrated an inhibitory effect of an extracellular, non-pathogenic resident gut microbe.
To test the role of autophagy in LPS-mediated inhibition of lysozyme, we tested the effects of rapamycin, an inducer of autophagy. Rapamycin was successful in countering the inhibitory effects of LPS on lysozyme production, demonstrating that autophagy is a positive regulator of lysozyme expression.
LPS may induce autophagy in RAW cells via a TLR4-dependent pathway. 16 We also found a moderate induction of autophagy by LPS in our study which is consistent with the previous findings (Figure 2c). However, this increase from LPS alone was insufficient to counter the inhibition of lysozyme by LPS in the absence of the stronger inducer of autophagy. One possible explanation for this is that LPS also induces NF-κB and NF-κB signaling can inhibit autophagy.24,25 Since rapamycin has been shown to inhibit NF-κB activation,26–28 this may explain the successful overcoming of inhibitory effects of LPS by rapamycin-induced autophagy in our study.
Triggering of autophagy alone, however, is not enough to affect lysozyme since we did not observe any effect on lysozyme expression when cells were treated with rapamycin alone (Figure 3a). LPS and autophagy are intimately linked in such a way that the full defensive role of autophagy is only revealed in the presence of the inhibitory effects of LPS. Our findings are in agreement with a published study which showed that adiponectin treatment counteracted LPS-induced TNF-α expression via autophagy induction. 29 In another published study, treatment of human monocytes with vitamin D and LPS, but not LPS alone, caused upregulation in genes of autophagy and antimicrobial peptide pathways. 30 These findings support the idea that induction of autophagy can overcome LPS-meditated effects and this is independent of autophagy triggered by LPS treatment alone.
Suppression of lysozyme expression by LPS and countering this effect by autophagy is clinically significant. One important example is that of a life-threatening Gram-negative sepsis, a major cause of mortality in hospitalized patients. The lethality of this condition may be understood in terms of a failure of autophagy on LPS-mediated inhibition of host defense. Since lysozyme decreases the toxicity of LPS and has an anti-septic effect,31–34 inhibition of lysozyme by LPS would favor the bacterial infection. We found not only that the prior treatment with rapamycin conferred protection against LPS-induced inhibition of lysozyme, but also reversed the effects of LPS on lysozyme expression when rapamycin was added after LPS treatment. Our findings suggest the therapeutic potential of both preventing and treating sepsis via induction of autophagy. Thus, our findings provide an explanation for the reported protective role of autophagy in sepsis.35,36 This protective effect of autophagy may be the result of its successful over-ride of endotoxin-mediated suppression of host production of AMPs. In contrast, lethal sepsis occurs when autophagic response is impaired. Our findings would explain why polymorphism in autophagy genes IRGM and ATG16L1 have been associated with susceptibility to sepsis.37,38
The loss of intracellular lysozyme is not due to increased secretion as confirmed by our finding that levels of lysozyme in the culture supernatant treated with LPS was comparable with control and in cells treated with rapamycin prior to LPS challenge. While our data show that lysozyme is inhibited at transcription and translation levels, it remains to be determined whether lysozyme is degraded by intracellular pathways in the presence of LPS.
Our data showed lytic activity of the culture supernatant against DSV, which was reduced by LPS in association with a significant decrease in lysozyme protein expression. We found that induction of autophagy by rapamycin protected against this effect. The observed lytic activity of lysozyme against DSV suggests that lysozyme activation downstream of autophagy may function to control the load of these extracellular resident gut bacteria and that deficiency in autophagic machinery may lead to decrease in lysozyme, which, in turn, may allow overgrowth of DSV in disease conditions.
The mechanism by which changes in the gut bacteria signal autophagic machinery and subsequent AMP production may also hold the key to potential treatment options for disorders linked to dysbiosis. In conclusion, our findings suggest that the role of autophagy in controlling microbes may be larger than our current understanding of its role as antimicrobial defense against intracellular pathogens.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Winkler Bacterial Overgrowth Research Fund.